 2014, Vol. 16
2014, Vol. 16



早期癫癎性脑病(early-onset epileptic encephalopathies,EEEs)是指发生在新生儿或婴儿 早期由反复临床惊厥或发作间期严重癎样放电导 致认知、运动全面落后、重度智力障碍等灾难性 神经系统后遗症的一类综合征[1]。国际抗癫癎联 盟(International League Against Epilepsy,ILAE)确 认的EEEs 包括大田原综合征(Ohtahara syndrome, OS)、婴儿痉挛症(West syndrome,WS)、婴儿 严重肌阵挛癫癎(Dravet syndrome)、早期肌阵挛 脑病、非进展性的肌阵挛脑病和婴儿严重局灶性 游走性癫癎。排除获得性脑损伤、脑结构异常、 遗传代谢性疾病、染色体病及单基因病等原因, 目前仍有半数病因不明。癫癎性脑病是一种遗传 异质性很高的症候群,且临床表型谱宽泛。至今 已明确EEEs 致病基因有18 个,部分基因通过全 基因组拷贝数变异(copy number variations,CNVs) 分析定位,全基因组CNVs 是指与基因组参考序列 相比数百bp 至数Mb 的微缺失/ 微重复[2]。目前 研究较多的是CNVs 与智力障碍、自闭症、精神 分裂症的关系[3];自2009 年以来,Helbig 等[4] 及 De Kovel 等[5] 发现全面性/ 部分性癫癎及癫癎脑病 与CNVs 密切相关,其中可能与多种全面性及局 灶性癫癎相关的潜在致病性CNVs 包括15q11.2、 16p11.2、6p13.11 等区域中的CNVs;Mefford 等 [6] 于2011 年对315 例癫癎性脑病的患者进行全基 因组CNVs 分析提示,7.9%(25/315) 患者携带 的CNVs 可能与其临床表型相关,并且发现了新 的癫癎性脑病候选基因:CACNA2D1、SLC1A3、 ADAM23。Bedoyan 等[7] 也发现16p11.2 的CNVs 与婴儿严重局灶性游走性癫癎相关;以上研究均 表明CNVs 是导致不明原因EEEs 的重要原因,目 前国内尚未见EEEs 全基因组CNVs 的分析报道。 本研究将从临床特点和全基因组CNVs 分析两方面 对不明原因EEEs 展开探讨。 1 资料与方法 1.1 研究对象
2012 年7 月至2013 年4 月我院小儿神经专科 门诊及住院部诊断为不明原因EEEs 患儿,纳入标 准:1 岁以内起病,临床表现为癫癎;伴以下条件 之一:(1)新生儿不明原因的痉挛、耐药癫癎、癫癎持续状态;(2)癫癎伴精神运动发育停滞或 倒退、运动障碍、头围改变、步态异常;(3)国 际抗癫癎联盟确认的可以在1 岁内起病的EEEs; (4)特殊的脑电图改变(爆发- 抑制、高度失 律、发作间期弥漫性慢波背景伴癫癎样放电持续 发放);⑸头颅MRI 异常(胼胝体发育不良、脑 萎缩等)。
排除标准:(1)血串联质谱和(或)尿气 相色谱- 质谱检查提示代谢病;(2)获得性脑损 伤:缺血缺氧性脑病、脑血管病变、外伤、颅内 感染、宫内感染等;(3)孤立的皮层发育异常; (4)染色体病及临床表型明确的单基因病。 1.2 资料采集
回顾性分析60 例不明原因EEEs 临床资料, 包括家族史、母孕史、癫癎起病年龄、发作形式、 精神运动发育评估、头围、血氨、血乳酸、脑电图、 颅脑影像学表现、治疗及预后。 1.3 全基因组CNVs 检测
经患儿家属同意后采集患儿及父母外周血 5 mL,EDTA 抗凝;采用血液基因组DNA 提取试 剂盒(天根公司)说明书操作提取DNA。全基因 组CNVs 检测使用Illumina 公司的SNP array 芯片 系统平台,HumanCyto-SNP12 Beadchips 包含有30 万个SNP 探针位点,平均每张芯片上每个探针的 重复数可高达30 次,探针分布覆盖245 种疾病 位点,HumanOmni-zhonghua 包含90 万个SNP 探 针位点。具体实验流程参照文献[8]。所得数据经 正常人群数据库DGV 数据库(http://projects.tcag. ca/variation),异常表型数据库(https://decipher. sanger.ac.uk)在线孟德尔遗传性疾病数据库OMIM (www.ncbi.nlm.nih.gov/omim) 以及PUBMED 分析 是否有已知致病性CNVs,对致病性或可疑致病 性CNVs 则行FISH 检测,并对父母进行检测做来 源分析,通过以上综合分析得出致病性CNVs。 目前认为:(1)新发的CNVs 位于已知的微缺失 微重复综合征区域内,或者与相关的综合征临床 表型相符考虑致病;(2)CNVs 片段的断裂点处 打断某些已知的致病性基因,且其临床表型符合 相关基因突变的临床表型时考虑致病;(3)新发 的>3 Mb 的CNVs 片段是致病的[8];(4)新发的 微缺失是致病的;(5)新发的重复可疑致病; (6)>1 Mb 的变异无论是否遗传或来源不明均可认为可疑致病[6]。(7)在正常人群的CNVs 研究中, 3 个以上的研究小组均发现有该CNVs 的记录,可 以认为是多态性改变。 2 结果 2.1 临床特点
60 例不明原因EEEs 患儿中,男44 例,女16 例,起病年龄为2 d 至1 岁,3 个月内起病21 人, 4~6 个月起病26 人,7~12 个月起病13 人,平均 起病年龄5.1±3.3 个月。根据临床表现及脑电图 特点临床诊断:WS 34 例(57%),OS 3 例(5%), Dravet 综合征3 例(5%),余20 例(33%)分型 不明确。
60 例患儿中1 例为领养儿,余59 例家族中有 癫癎和(或)智力低下者8 人。母亲既往有自然 流产史者4 例,死胎史者1 例;母孕期间有先兆 流产表现12 例。
34 例WS 患儿表现为痉挛发作,6 例合并强 直或强直- 阵挛发作,1 例合并部分运动性发作; 3 例OS 患儿表现为痉挛发作,其中1 例合并强直- 阵挛发作;3 例Dravet 综合征患儿以肌阵挛、强直 阵挛发作为主,可伴部分性发作;20 例分型不明 确EEEs 患儿中8 例为强直或强直阵挛发作;5 例 为肌阵挛发作;3 例为多种发作形式(强直- 阵挛 并部分运动性发作或痉挛发作);2 例为痉挛发作; 2 例为部分性运动发作。
根据世界卫生组织(WHO)和美国智力低下 协会(AAMD)的智力低下分级标准[9],轻度智力 缺损(IQ 50~69)14 人(23%),中度智力缺损(IQ 35~49)22 人(37%),重度智力缺损(IQ 34 以下) 24 人(40%)。
共统计了54 例患儿头围,其中(x + s ~ x + 2 s) 2 例(4%),(x ~ x+s)3 例(6%),(x-s ~ x) 11 例(20%),(x-2 s ~ x-s)11 例(20%), (x-3 s ~ x-2 s)18 例(33%),≤ x-3 s )9 例(17%)。
51 例患儿检测了血氨(参考值10~47 μmol/L), 平均44±24 μmol/L,其中15 人(29%)血氨升高 ( 平均68±30 μmol/L),最高值144 μmol/L;46 人检测了血乳酸(参考值0.5~2.22 mmol/L),平均 2.8±1.4 μmol/L,其中19 人升高(3.8±0.93 mmol/L), 最高值6.4 mmol/L。
34 例WS 患儿脑电图改变为发作间期高度失 律或间断高度失律,其中1 人醒睡各期或非快速 眼动期(non-rapid eye movement,NREM)放电指数 达95%;3 例OS 患儿脑电图表现为睡眠期间断爆 发- 抑制,1 例醒睡各期或NREM 期放电指数达 95%。3 例Dravet 综合征患儿中,2 例提示全导棘 慢波、多棘慢波发放,1 例为局灶尖棘波发放;20 例分型不明确不明原因EEEs 患儿4 例提示高度失 律,4 例全导棘波、棘慢波、多棘慢波爆发。4 例 提示电背景活动慢化,4 例为多灶性尖波、尖棘波 发放,其中2 例监测到部分运动性发作;2 例表现 为爆发- 抑制,1 例为发作期全导低电压,1 例为 全导慢波阵发。
60 例不明原因EEEs 患儿6 例完善头部CT 平 扫或平扫增强,54 例完善颅脑MRI 平扫或平扫增 强,其中11 例提示脑外侧裂增宽或外部性脑积水, 4 例侧脑室增宽,胼胝体发育不良3 例,3 例脑白 质异常信号,2 例透明隔囊肿,3 例蛛网膜下囊肿, 34 例无明显异常。
34 例WS 患儿中27 例采用ACTH 冲击治疗联 合1~3 种抗癫癎药物,2 例采用2~3 联抗癫癎治疗, 1 例单药治疗,20 例癫癎已控制,12 例未控制, 2 例死亡。20 例未分型癫癎脑病患儿9 例单药治 疗,11 例2~3 种抗癫癎药物联合治疗,8 例癫癎 已控制,8 例未控制,3 例死亡,失访1 例。3 例 Dravet 综合征患儿予3 联抗癫癎治疗、3 例OS 患 儿予ACTH 联合1~2 种抗癫癎药物治疗均未控制。 2.2 全基因组CNVs 分析结果
(1) 全基因组CNVs 结果:60 例不明原因 EEEs 患儿中,5 例患儿携带7 个致病性或可疑致 病性CNVs,阳性率为8%,具体位点见表 1。
| 表 1 致病性或可疑致病性CNVs 详情 |
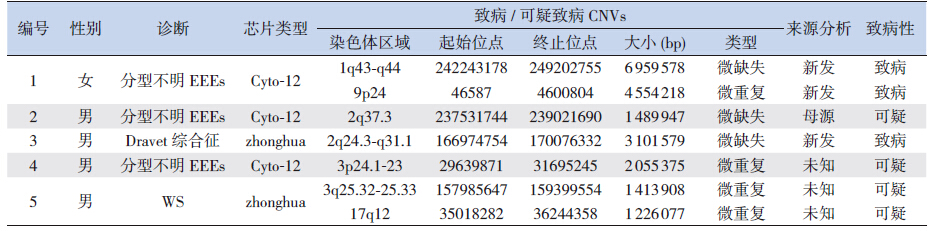
(2) 基因型表型分析:① 1q43q44 微缺失 可导致一系列已明确临床症状,包括不同程度的 智力障碍、语言发育迟缓、面部及手足异常、小 头畸形、胼胝体发育不良、癫癎[10],与患儿1 临 床表现相符。经FISH 验证及父母来源分析确定 为1q43q44 微缺失合并9p24 微重复新发突变, 致病性明确,且为首次报道。②异常表型数据 库265 334 患者2q37.3 出现0.15 Mb 微缺失,表 现为智力低下、多动及面容异常,表明2q37.3 可 能与智力障碍及癫癎相关,为可能致病性突变。 ③ 2q24.3-q31.1 微缺失部分包括17 个基因,其中包括部分SCN1A:电压门控钠离子通道α 亚单位 神经亚型Ⅰ型(OMIM:182389),为Dravet 综合 征致病基因[11, 12],符合患儿临床诊断,经FISH 验 证及父母来源分析为新发突变,致病性明确。缺 失部分还包括 SCN9A(OMIM:603415)、SCN7A (OMIM:182392),Singh 等 [13] 认为SCN9A 对 Dravet 综合征有修饰作用,而Gorter 等 [14] 发现癫 癎大鼠模型及癫癎患者海马区SCN7A 基因的持续 表达可能与颞叶癫癎的进展相关,因此SCN9A、 SCN7A 与Dravet 综合征的关系值得进一步探讨。 ④ 3p24.1-23 微重复片段共包含5 个非癫癎相关基 因。异常表型数据库275459 患儿3p24.1 的微重复 与精神异常相关;Chioza 等 [15] 经全基因组连锁分 析证实3p23-p14 与儿童失神癫癎相关。⑤异常表 型数据库285 007 患儿3q25.32 存在0.15 Mb 微重复, 与智力障碍及肥胖有关。异常表型数据库多个患 者(282150、281079、278411 等)17q12 的微重复 与智障、癫癎、小头畸形相关,提示为致病性变 异可能性大。 3 讨论
本研究60 例不明原因EEEs 患儿中,男性病 例较多,男女比例为2.75 : 1,且多于新生儿期及 婴儿早期起病,平均发病年龄5±3 个月,78% 在 6 个月以内起病。在所有已知EEEs 中以婴儿痉挛 症发病率最高,为0.16~0.45‰ [16, 17],本组患儿中, 婴儿痉挛症(WS)最常见,占57%,而分型不明 确者占33%。已知综合征患儿临床及脑电图表现 典型,分型不明确的不明原因EEEs 患儿癫癎发作 形式以强直/ 强直- 阵挛、肌阵挛及痉挛发作为主, 脑电图表现多样,包括高度失律、全导棘波、尖棘 波、棘慢波爆发,电背景活动慢化、爆发- 抑制等。由于其临床表现多样,脑电图表现无规律可循,目 前对分型不明确癫癎性脑病报道甚少,加强对该类 患儿临床特点总结及遗传病因学诊断,有助于提高 不明原因EEEs 的诊疗水平。
51 例检测了血氨水平的患儿中15 例(29%) 血氨升高;46 例检测了血乳酸的患儿中,19 例 (41%)血乳酸升高,提示不明原因EEEs 患儿存 在不同程度的代谢异常。54 例检测了头围的患儿 中,49 例(91%)头围位于同性别同龄均值及以 下,小头畸形占总人数的17%。35% 患儿颅脑影 像学检查提示脑发育不良或脑萎缩。经抗癫癎治疗 后47% 患儿癫癎已控制,但76% 患儿伴有中重度 智力障碍,随访1~1.5 年有5 例死亡,说明不明原 因EEEs 致残致死率高。
如何明确不明原因EEEs 病因,通过提高产 前诊断水平减少此类患儿出生是目前亟待解决的 问题。本研究是国内首次对不明原因EEEs 行全 基因组CNVs 筛查,阳性率达8%(5/60),与 Mefford 等 [6] 报道相仿。5 例患儿携带可疑或致病 性CNVs,涉及区域有1q43-q44、9p24、2q37.3、 2q24.3-q31.1、3p24.1-23、3q25.32-25.33、17q12, 未包含Mefford HC 团队已报道的癫癎脑病热点 CNVs:15q13.3、16p13.11、15q11.2,可能与样本 量少或种族差异有关。全基因组CNVs 分析费用 低、周期短,在有效检测微缺失/ 重复的同时还可 定位新的EEEs 候选基因,但遗憾的是由于本组样 本量较少,CNVs 阳性结果中无重复变异片段,因 此未发现新的EEEs 候选基因。本研究通过全基因 组CNVs 分析丰富了不明原因EEEs 基因型,提高 了对不明原因EEEs 的病因诊断水平,为患儿家庭 再生育的遗传咨询提供了理论依据,是一种值得推
| [1] | Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies:Report of the ILAE Commission on Classification and Terminology, 2005-2009[J]. Epilepsia, 2010, 51(4):676-685. |
| [2] | Feuk L, Carson AR, Scherer SW. Structural variation in the human genome[J]. Nat Rev Genet, 2006, 7(2):85-97. |
| [3] | Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease[J]. Curr Opin Genet Dev, 2009, 19(3):196-204. |
| [4] | Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy [J]. Nat Genet, 2009, 41(2):160-162. |
| [5] | De Kovel CG, Trucks H, Helbig I, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies[J]. Brain, 2010, 133(Pt 1):23-32. |
| [6] | Mefford HC, Yendle SC, Hsu C, et al. Rare copy number variants are an important cause of epileptic encephalopathies [J]. Ann Neurol, 2011, 70(6):974-985. |
| [7] | Bedoyan JK, Kumar RA, Sudi J, et al. Duplication 16p11.2 in a child with infantile seizure disorder[J]. Am J Med Genet A, 2010, 152A(6):1567-1574. |
| [8] | 刘静. 373 例不明原因智力障碍/生长发育迟缓,多发畸形 患者的全基因组拷贝数变异分析[D]. 中南大学, 2013. |
| [9] | 何慧静, 万国斌, 韦臻. 223 例智力障碍儿童的病因分析[J]. 中国优生与遗传杂志, 2010, 18(6):142-143. |
| [10] | Ballif BC1, Rosenfeld JA, Traylor R, et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44[J]. Hum Genet, 2012, 131(1):145-156. |
| [11] | Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy[J]. Am J Hum Genet, 2001, 68(6):1327-1332. |
| [12] | Depienne C, Trouillard O, Saint-Martin C, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome:analysis of 333 patients[J]. Med Genet, 2009, 46(3):183-191. |
| [13] | Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome[J]. PLoS Genet, 2009, 5(9):e1000649. |
| [14] | Gorter JA, Zurolo E, Iyer A, et al. Induction of sodium channel Na(x) (SCN7A) expression in rat and human hippocampus in temporal lobe epilepsy[J]. Aronica E Epilepsia, 2010, 51(9):1791-1800. |
| [15] | Chioza BA, Aicardi J, Aschauer H, et al. Genome wide high density SNP-based linkage analysis of childhood absence epilepsy identifies a susceptibility locus on chromosome 3p23-p14[J]. Epilepsy Res, 2009, 87(2-3):247-255. |
| [16] | Covanis A. Epileptic encephalopathies(including severe epilepsy syndromes) [J]. Epilepsia, 2012, 53(Suppl 4):114-126. |
| [17] | Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms:AU.S. Consensus report[J]. Epilepsia, 2010, 51(10):2175-2189. |