 2014, Vol. 16
2014, Vol. 16



Alagille 综合征(Alagille syndrome,ALGS)是 一种常染色体显性遗传病,临床表现主要包括胆 汁淤积和胆管稀疏、先天性心脏病、面部异常、 蝴蝶椎以及眼部异常,还有肾脏异常、生长发育 迟缓、胰腺异常等[1]。随着家系研究和基因检测的 开展,近年来报道病例逐渐增多,特定人群患病 率可达1 : 30 000[2]。94% 的ALGS 由编码JAGGED1 的JAG1 基因突变或缺失所引起,约1.5% 由 NOTCH2 基因突变导致,但有4.5%未检测到基因 突变[3]。JAG1 和NOTCH2 基因都是Notch 信号转 导通路的重要组成部分,这两个基因的新突变多 发[4],给ALGS 确诊带来一定困难。虽然国外已经 有大量ALGS 研究报道,但国内有关本病的正式文 献尚不多见。本文对ALGS 诊治进展进行系统全面 的总结,希望能为本病的诊断和治疗提供参考。 1 病因及机制 1.1 JAG1 基因
JAG1 基因定位在染色体20p12,编码细胞膜 表面蛋白JAGGED1。JAGGED1 是Notch 受体的功 能性配体,受体与配体相互作用启动下游信号转 录,从而影响细胞的增殖与分化[5]。生长发育过程 中JAG1 在心血管系统,特别是在全身动脉中表达。 在体外,Notch 信号控制细胞增殖和血管内皮细胞 的迁移和分化;在体内,Notch 信号通路促进心脏 中上皮- 间质细胞转型,诱导血管的生成,并且 Notch 信号可通过促进心肌再生、保护缺血心肌和抑制心脏成纤维细胞- 肌成纤维细胞转化来修复 心肌损伤[6]。在越南ALGS 患者中90% 可检测到 JAG1 突变,且80%(17/21)都是未被报道的新突 变[7],说明JAG1 基因突变具有高度异质性。 1.2 NOTCH2 基因
McDaniell 等[8] 筛选了11 例JAG1 突变阴性 的ALGS 患者,发现了NOTCH2 基因的突变。 NOTCH2 基因在近端肾单位的形成中起重要作用, 其突变可导致肾发育不良及蛋白尿[9]。该基因突变 的ALGS 患者多具有胆管稀疏,但很少发现骨骼畸 形及面部特征性改变,不完全符合传统诊断标准[10]。 1.3 发病机制
目前研究发现Notch 信号在肝内胆管(IHBDs) 的生成及维持中起重要作用[11]。Notch 信号缺乏 导致肝内胆管生成异常,胆管内皮细胞减少,并 导致肝内胆管的主分支及中间支生成异常。RBP (Notch 信号的组成部分)缺乏小鼠的单位门静脉 所含胆管减少,且单位汇管区所含门静脉减少[12]。
Notch 信号在心血管系统发育及稳态维持中起 重要作用,JAGGED1 在胚胎期即有表达,特别是 在血管内皮细胞。Notch 信号缺失将会导致右心室 肥大、肺动脉狭窄、室间隔缺损、冠状动脉异常 及瓣膜缺损[13]。在心内膜垫的形成过程中,JAG1 的缺失将会破坏内皮细胞向间充质转化,影响心 内膜垫的形成。JAG1 突变成年小鼠表现出与异常 基质重塑相关的心脏瓣膜钙化[14]。
Zanotti 等[15] 发现Notch 信号可以调节骨骼发 育和重塑。Notch 信号缺乏不仅导致骨骼发育障碍 和骨质流失,而且在骨肉瘤的发展和乳腺癌的骨 转移方面也有促进作用。
Notch 信号对近端肾小管上皮细胞以及肾集合 管系统的的发育起重要作用,并且对损伤修复及 组织稳态也起关键作用,急性肾损伤的非ALGS 病 人Notch 信号表达升高以启动修复机制[16]。此外, 胆汁淤积还可以使载脂蛋白A-I、HDL、VLDL 等 合成障碍,引起高脂血症,从而引发肾脏脂质沉积, 引发系膜增生性肾小球肾炎、微小病变性肾小球 肾炎等[17]。
颅面受累机制的报道较少,Humphreys 等[18] 通过研究在颅面发育中起重要作用的颅神经嵴细 胞(CNS 细胞),发现Jagged1 敲除的小鼠CNS 细胞增殖减少,细胞基质减少、分支血管生成减少,从而导致中面部发育不良,30 d 后小鼠死于下颚 错位及口腔闭塞导致的无法咀嚼。
眼部受累机制的报道较为罕见,眼睛受累后 可表现为视乳头水肿,Yilmaz 等[19] 的研究发现 Jagged1 可参与颅缝闭合,ALGS 的患儿颅缝早闭, 怀疑颅压升高导致视乳头水肿。 2 临床表现 2.1 肝脏
在生后3 个月内甚至新生儿期大部分患儿即 开始出现胆汁淤积并逐渐发展,出现黄疸、皮肤 瘙痒、白陶土样大便及高脂血症,尤其以血中胆 固醇升高最明显。皮肤瘙痒的症状可能比黄疸更 明显,约33% 的患儿会出现瘙痒[20]。肝病早期 仅表现为轻度肝酶水平异常,白球比倒置较为少 见,可进展为进行性黄疸、轻度急性自限性肝炎样 疾病、自身免疫型肝炎、暴发性肝功能衰竭或慢 性肝脏疾病等,但很少发生肝硬化[21]。肝脾肿大 见于多数ALGS 患儿。一般血中胆红素会有明显 升高,可达正常上限的10~30 倍,胆汁酸可能会 更高。Kamath 等[22] 曾报道,5 岁前总胆红素高于 6.5 mg/dL,结合胆红素高于4.5 mg/dL 和胆固醇高 于520 mg/dL 的患儿都有可能进展为严重肝病,而 低于该数值的患儿预后相对良好。 2.2 心脏
ALGS 病人中多数出现心脏杂音,多由肺动脉 流出道狭窄所引起。肺动脉病变多单发,也可与 其他心脏病变同时出现,其中周围肺动脉和肺动 脉瓣狭窄占67%,法洛四联症占16%,其他畸形 包括室间隔缺损、房间隔缺损、主动脉瓣狭窄和 主动脉缩窄等[13],心血管异常发育的严重程度也 与患儿预后有关[21]。 2.3 骨骼
ALGS 病人可出现骨骼发育障碍和骨质流失, 具体表现为脊柱畸形,X 线可见蝶形椎骨,偶尔 可见椎体融合、隐性脊柱裂等。蝶形椎骨并非 见于每个患者,Wang 等[23] 研究显示其发生率约 46%。骨骼异常通常无显性症状,一般在X 线检 查时发现。除脊柱病变外,少数患者出现四肢骨 骼病变,多表现为骨质疏松或骨质缺失,如上下 肢缩短、浮肋缺如、股骨病理性骨折等[24]。 2.4 面部
ALGS 患者可出现典型面部畸形,如前额宽 阔、眼窝深陷、耳廓突出、眼距增宽、尖下巴, 整张脸犹如一个三角形,呈V 字形[18]。上述特征 在婴幼儿期可不明显,而随着年龄增长逐渐显现 出来。早期研究认为ALGS 的面部特征无特异性, 但Kamath 等[25] 认为与其他形式的先天性肝内胆 汁淤积症导致的面部异常相比,ALGS 的面部畸形 的特异度为79%。 2.5 眼部
眼部异常以角膜后胚胎环(Schwalbe's 环)最 常见,其发生率约占ALGS 病人的90%,多发生 于角膜内皮和葡萄膜(虹膜)小梁网[26]。此外, 由于ALGS 影响角膜、结膜、视网膜、视神经盘 等,Makino 等[27] 发现了首例视网膜萎缩累及黄斑 的ALGS 患儿,因此各种眼科症状都有可能发生。 约13% 的病人可以看到阿克森费尔德异常,即青 光眼及角膜巩膜发育不全,会出现圆锥角膜、先 天性黄斑营养不良、前房浅、外斜视、带状角膜 病和白内障等。 2.6 其他
除以上几大主要表现外,一些其他器官的临 床表现也与ALGS 有关,其中肾病倍受关注,约 40% 的ALGS 患者合并肾脏受累,具体表现为肾 小管性酸中毒、肾发育不良、蛋白尿肾囊肿、尿 路梗阻等[8]。ALGS 也可导致生长发育障碍、运动 迟缓、胰腺功能不全等[28, 29]。此外,口腔健康依 赖于肝脏的疾病状态[30],牙科表现并非ALGS 的 主要特征,但它们可作为胆汁长期淤积的一种并 发症。胆汁淤积可致牙釉质混浊、矿质过少和牙 齿的色素沉着等[31]。 3 诊断
经典的ALGS 诊断标准为同时满足慢性胆汁 淤积、心脏疾病、骨骼异常、眼部异常和面部特 征等五大临床表现[32],具体见表 1。其他临床表现 包括肾功能异常[33]、生长发育迟缓和智力发育落 后也有助于诊断ALGS[34]。
| 表 1 经典的ALGS 征诊断标准 |
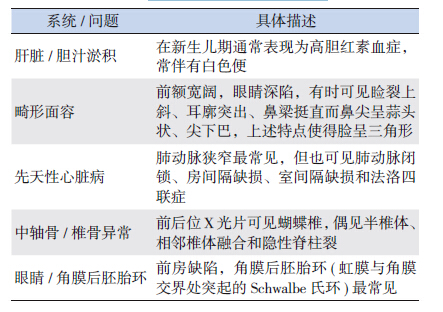
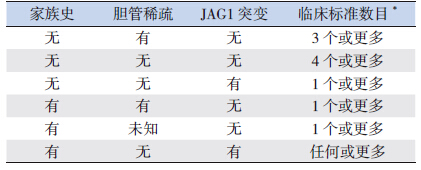
值得注意的是,并非所有ALGS 患者都同时 满足上述5 条典型诊断标准[35],有国外文献认为 符合上述标准中的3 条即可诊断ALGS[36, 37]。虽然过于严格的诊断标准可能导致漏诊,但是标准过 低则可能导致误诊误治。作为一种常染色体显性 遗传病,综合考虑临床表现、家族史、肝脏病理 改变和基因突变等依据对于ALGS 诊断可能是必要 的。因此,Kamath 等[38] 和Guru 等[39] 提出了修订 的AGLS 诊断标准(表 2)。该修订标准强调了肾 脏异常和JAG1 基因突变对于ALGS 的诊断价值, 但随着分子生物学研究的进展,NOTCH2 突变也 应被考虑[40]。
| 表 2 修订的ALGS 诊断标准 |
ALGS 目前尚无病因治疗措施,其治疗以对症 支持为主。对于诊断为ALGS 的病人,应注意监测 各个器官的功能[41]。良好的营养可改善生长发育 落后[40],摄取多种食物的同时,要注意食物之间 的搭配,做到平衡膳食。除补充适当的糖、脂肪、 蛋白质外,还应注意补充微量元素及脂溶性维生素[42]。他汀类药物( 包括洛伐他汀、氟伐他汀、 普伐他汀、辛伐他汀、阿托伐他汀以及最近的瑞 舒伐他汀等) 治疗,可以有效降低患儿总胆固醇和 低密度脂蛋白胆固醇[43],但其远期疗效有待观察。
眼部症状多对症治疗,Fukumoto 等[44] 对 ALGS 相关白内障病人进行白内障超声乳化术及人 工晶体植入术,缓解了眼部症状。肾脏损伤可考 虑透析治疗,但对于严重肾病可进行肾移植[45]。
ALGS 患儿的最终预后取决于肝脏和心脏疾病 的严重程度[21]。对于肝病引起的严重的皮肤瘙痒 可使用阿片受体拮抗剂纳曲酮、消胆胺和利福平 进行治疗[40],若无改善可考虑肝移植[46]。Kamath 等[47] 发现ALGS 的患儿肝移植后的存活率显著低 于先天性胆道闭锁的患儿。Lee 等[48] 对9 例ALGS 儿童进行肝移植手术,活体肝移植后的5 年期和 20 年总生存率分别为88.9%和77.8%。患者移植 后随访4.3~25.7 年不等,移植后第30 天因肝动脉 血栓形成死亡1 例,另1 例死于消化道出血和颅 内出血并发脑疝。
周围肺动脉狭窄是ALGS 最常见的心脏异常, 大多数中心提倡进行导管介入干预,疗效较好, 但多需要再次介入治疗。Cunningham 等[49] 对69 例行肺动脉介入治疗的肺动脉狭窄儿童进行分析, 发现初次干预后平均右心室/ 左心室压比值从1.00 下降至0.88,再次介入后可下降至0.53。随访 1 年后有(38±6)% 的病人不需再次介入,5 年 后下降至(22±6)%,有18 例病人接受了3 次 以上的治疗。在平均8.5 年的随访中死亡10 例, 出现并发症5 例,但均为1998 年以前的病例。 有证据表明,外科手术也能获得较好的疗效, Mainwaring 等[50] 对2001~2011 年期间接受外科肺 动脉重建术的患儿进行分析,没有发现早期及远 期死亡,随访时间为11 个月到9 年。Monge 等[51] 对16 例行肺动脉重建术的患者进行分析,术中死 亡1 例,右心室/ 左心室压力比值下降至0.40,比 术前下降了55%,在5 年的随访中,并未发现死 亡及需要再次手术的情况,因此认为肺动脉重建 术有利于患者长期生存。
| [1] | Ciocca M, Alvarez F. Alagille syndrome[J]. Arch Argent Pediatr, 2012, 110(6):509-515. |
| [2] | Kamath BM, Yin W, Miller H, et al. Outcomes of liver transplantation for patients with Alagille syndrome:the studies of pediatric liver transplantation experience[J]. Liver Transpl, 2012, 18(8):940-948. |
| [3] | Leonard LD, Chao G, Baker A, et al. Clinical utility gene card for:Alagille syndrome (ALGS)[J]. Eur J Hum Genet, 2014, 22(3):e1-e4. |
| [4] | Vozzi D, Licastro D, Martelossi S, et al. Alagille syndrome:a new missense mutation detected by whole-exome sequencing in a case previously found to be negative by DHPLC and MLPA[J]. Mol Syndromol, 2013, 4(4):207-210. |
| [5] | Vanorny DA, Prasasya RD, Chalpe AJ, et al. Notch signaling regulates ovarian follicle formation and coordinates follicular growth[J]. Mol Endocrinol, 2014, 28(4):499-511. |
| [6] | Zhou XL, Liu JC. Role of Notch signaling in the mammalian heart[J]. Braz J Med Biol Res, 2014, 47(1):1-10. |
| [7] | Lin HC, Le Hoang P, Hutchinson A, et al. Alagille syndrome in a Vietnamese cohort:mutation analysis and assessment of facial features[J]. Am J Med Genet A, 2012, 158A(5):1005-1013. |
| [8] | McDaniell R, Warthen DM, Sanchez-Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway[J]. Am J Hum Genet, 2006, 79(1):169-173. |
| [9] | Kamath BM, Spinner NB, Rosenblum ND. Renal involvement and the role of Notch signalling in Alagille syndrome[J]. Nat Rev Nephrol, 2013, 9(7):409-418. |
| [10] | Kamath BM, Bauer RC, Loomes KM, et al. NOTCH2 mutations in Alagille syndrome[J]. J Med Genet, 2012, 49(2):138-144. |
| [11] | Sparks EE, Huppert KA, Brown MA, et al. Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice[J]. Hepatology, 2010, 51(4):1391-1400. |
| [12] | Sparks EE, Perrien DS, Huppert KA, et al. Defects in hepatic Notch signaling result in disruption of the communicating intrahepatic bile duct network in mice[J]. Dis Model Mech, 2011, 4(3):359-367. |
| [13] | Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease[J]. Semin Cell Dev Biol, 2012, 23(4):450-457. |
| [14] | Hofmann JJ, Briot A, Enciso J, et al. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome[J]. Development, 2012, 139(23):4449-4460. |
| [15] | Zanotti S, Canalis E. Notch regulation of bone development and remodeling and related skeletal disorders[J]. Calcif Tissue Int, 2012, 90(2):69-75. |
| [16] | Sirin Y, Susztak K. Notch in the kidney:development and disease[J]. J Pathol, 2012, 226(2):394-403. |
| [17] | Benoit G, Sartelet H, Levy E, et al. Mesangiolipidosis in Alagille syndrome-relationship with apolipoprotein A-I[J]. Nephrol Dial Transplant, 2007, 22(7):2072-2075. |
| [18] | Humphreys R, Zheng W, Prince LS, et al. Cranial neural crest ablation of Jagged1 recapitulates the craniofacial phenotype of Alagille syndrome patients[J]. Hum Mol Genet, 2012, 21(6):1374-1383. |
| [19] | Yilmaz S, Turhan T, Mutluer S, et al. The association of Alagille syndrome and craniosynostosis[J]. Pediatr Neurol, 2013, 48(2):146-148. |
| [20] | Srivastava A, Goel D, Bolia R, et al. Alagille syndrome:experience of a tertiary care center in North India[J]. Indian J Gastroenterol, 2014, 33(1):59-62. |
| [21] | Vajro P, Ferrante L, Paolella G. Alagille syndrome:an overview[J]. Clin Res Hepatol Gastroenterol, 2012, 36(3):275-277. |
| [22] | Kamath BM, Munoz PS, Bab N, et al. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome[J]. J Pediatr Gastroenterol Nutr, 2010, 50(5):526-530. |
| [23] | Wang JS, Wang XH, Zhu QR, et al. Clinical and pathological characteristics of Alagille syndrome in Chinese children[J]. World J Pediatr, 2008, 4(4):283-288. |
| [24] | 王建设, 王晓红, 王中林, 等. Alagille 综合征五例临床和病 理特点[J]. 中华儿科杂志, 2007, 45(4):308-309. |
| [25] | Bales CB, Kamath BM, Munoz PS, et al. Pathologic lower extremity fractures in children with Alagille syndrome[J]. J Pediatr Gastroenterol Nutr, 2010, 51(1):66-70. |
| [26] | Kamath BM, Loomes KM, Oakey RJ, et al. Facial features in Alagille syndrome:specific or cholestasis facies? [J]. Am J Med Genet, 2002, 112(2):163-170. |
| [27] | Mozhgan Z, Bita G, Mahmood H, et al. Paucity of intrahepatic bile ducts in neonates:the first case series from iran[J]. Iran J Pediatr, 2013, 23(1):65-70. |
| [28] | Makino S, Ohkubo Y, Tampo H. Optical coherence tomography and fundus autofluorescence imaging study of chorioretinal atrophy involving the macula in Alagille syndrome[J]. Clin Ophthalmol, 2012, 6:1445-1448. |
| [29] | Kamath BM, Piccoli DA, Magee JC, et al. Pancreatic insufficiency is not a prevalent problem in Alagille syndrome[J]. J Pediatr Gastroenterol Nutr, 2012, 55(5):612-614. 29] Fouillade C, Monet-Leprêtre M, Baron-Menguy C, et al. Notch signalling in smooth muscle cells during development and disease[J]. Cardiovasc Res, 2012, 95(2):138-146. |
| [30] | Olczak-Kowalczyk D, Pawłowska J, Kowalczyk W. Oral health status in children with chronic liver disease[J]. J Stoma, 2011, 64(10):760-774. |
| [31] | Berniczei-Royko A, Chałas R, Mitura I, et al. Medical and dental management of Alagille syndrome:A review[J]. Med Sci Monit, 2014, 20:476-480. |
| [32] | Alagille D, Estrada A, Hadchouel M, et al. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia):review of 80 cases[J]. J Pediatr, 1987, 110(2):195-200. |
| [33] | Salem JE, Bruguiere E, Iserin L, et al. Hypertension and aortorenal disease in Alagille syndrome[J]. J Hypertens, 2012, 30(7):1300-1306. |
| [34] | Olsen IE, Ittenbach RF, Rovner AJ, et al. Deficits in sizeadjusted bone mass in children with Alagille syndrome[J]. J Pediatr Gastroenterol Nutr, 2005, 40(1):76-82. |
| [35] | Kamath BM, Bason L, Piccoli DA, et al. Consequences of JAG1 mutations[J]. J Med Genet, 2003, 40(12):891-895. |
| [36] | McElhinney DB, Krantz ID, Bason L, et al. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome[J]. Circulation, 2002, 106(20):2567-2574. |
| [37] | Witt H, Neumann LM, Grollmuss O, et al. Prenatal diagnosis of Alagille syndrome[J]. J Pediatr Gastroenterol Nutr, 2004, 38(1):105-106. |
| [38] | Kamath BM. Alagille syndrome[M]//Suchy FJ, Sokal RJ, Balistreri WF, et al. Liver Disease in Children.3rd ed. New York:Cambridge University Press, 2007:326-345. |
| [39] | Guru Murthy GS, Rana BS, Das A, et al. Alagille syndrome:a rare disease in an adolescent[J]. Dig Dis Sci, 2012, 57(11):3035-3057. |
| [40] | Turnpenny PD, Ellard S. Alagille syndrome:pathogenesis, diagnosis and management[J]. Eur J Hum Genet, 2012, 20(3):251-257. |
| [41] | Kamath BM, Loomes KM, Piccoli DA. Medical management of Alagille syndrome[J]. J Pediatr Gastroenterol Nutr, 2010, 50(6):580-586. |
| [42] | Moisseiev E, Cohen S, Dotan G. Alagille syndrome associated with xerophthalmia[J]. Case Rep Ophthalmol, 2013, 4(3):311-315. |
| [43] | Tapia Ceballos L, Picazo Angelín B, Ruiz García C. Use of statins in children[J]. An Pediatr (Barc), 2008, 68(4):385-392. |
| [44] | Fukumoto M, Ikeda T, Sugiyama T, et al. A case of Alagille syndrome complicated by intraocular lens subluxation and rhegmatogenous retinal detachment[J]. Clin Ophthalmol, 2013, 7:1463-1465. |
| [45] | Shrivastava R, Williams A, Mikhail A, et al. An unusual cause of hypertension and renal failure:a case series of a family with Alagille syndrome[J]. Nephrol Dial Transplant, 2010, 25(5):1501-1506. |
| [46] | Mozer-Glassberg Y, Hojsak I, Zevit N, et al. Pruritus responsive to naltrexone in a patient with cholestatic liver disease[J]. Isr Med Assoc J, 2011, 13(2):111-112. |
| [47] | Kamath BM, Yin W, Miller H, et al. Outcomes of liver transplantation for patients with Alagille syndrome:the studies of pediatric liver transplantation experience[J]. Liver Transpl, 2012, 18(8):940-948. |
| [48] | Lee CN, Tiao MM, Chen HJ, et al. Characteristics and outcome of liver transplantation in children with Alagille syndrome:a singlecenter experience[J]. Pediatr Neonatol, 2014, 55(2):135-138. |
| [49] | Cunningham JW, McElhinney DB, Gauvreau K, et al. Outcomes after primary transcatheter therapy in infants and young children with severe bilateral peripheral pulmonary artery stenosis[J]. Circ Cardiovasc Interv, 2013, 6(4):460-467. |
| [50] | Mainwaring RD, Sheikh AY, Punn R, et al. Surgical outcomes for patients with pulmonary atresia/major aortopulmonary collaterals and Alagille syndrome[J]. Eur J Cardiothorac Surg, 2012, 42(2):235-240. |
| [51] | Monge MC, Mainwaring RD, Sheikh AY, et al. Surgical reconstruction of peripheral pulmonary artery stenosis in Williams and Alagille syndromes[J]. J Thorac Cardiovasc Surg, 2013, 145(2):476-481. |