 2014, Vol. 16
2014, Vol. 16



2. 兰州军区乌鲁木齐总院临床医学研究所, 新疆 乌鲁木齐 830000
苯丙酮尿症(phenylketonuria,PKU) 是由于 脏苯丙氨酸羟化酶(phenylalanine hydroxylase, PAH)活性减低或缺乏,导致苯丙氨酸(phenylatanine, Phe)代谢障碍的一种常染色体隐性遗传病, 其主要致病原因是苯丙氨酸羟化酶(PAH)基因发 生突变[1]。研究发现,我国PKU 患者PAH 基因外 显子7 突变检出率最高[2]。宁夏位于我国西北地区, 人口以回族和汉族为主,其中回族约占1/3,PKU发病率为0.29‰(1/3 423)[3]。本研究对临床确诊 的73 例经典型PKU 患儿PAH 基因外显子7 进行 突变检测分析,探讨宁夏地区PAH 基因突变的特 点,为该地区开展PKU 产前基因诊断提供依据。 1 资料与方法 1.1 研究对象
73 例(回族39 例,汉族34 例)经典型PKU 患儿均为宁夏新生儿疾病筛查中心于2010 年1 月 至2012 年12 月确诊,其中64 例为新生儿筛查确诊, 9 例为出现症状后确诊。年龄15 d 至13 岁,男性 38 例(回族17 例),女性35 例(回族22 例)。 初诊血Phe 浓度在582~2 940 μmol/L 之间( 正常 参考值<120 μmol/L)。所有患儿均进行了尿喋呤 谱分析、血二氢蝶啶还原酶测定和四氢生物蝶呤 负荷实验,排除了四氢生物蝶呤缺乏症。另选取 100 名正常对照者(回族50 名,汉族50 名,均为 2012 年宁夏新生儿筛查标本),其血清Phe 浓度 <120 μmol/L,无PKU 家族史。本研究获得患儿家 长或监护人的书面知情同意。 1.2 研究方法 1.2.1 DNA 提取
应用血片卡快速酚/ 氯仿 抽提法,将带有血样的干滤纸卡片沿血滴4 周 剪下, 用1×TE500 μL+ NaCl(250 mmol/L) 800 μL 洗涤1 次, 加入200 μL 消化缓冲液 (100 mmol/L Tris pH 8.0,20 mmol/L ED-TA, 150 mmol/L NaCl,2% SDS),5 μL 蛋白酶K, 65℃水浴1 h。不去除纸片直接在消化液中加入 200 μL 饱和酚- 氯仿- 异戊醇(25 : 24 : 1)提取基 因组DNA。 1.2.2 PCR 扩增
PAH 基因外显子7 区域PCR 引物,严格按照引物序列设计原则设计完成[4],正 向引物序列:5'-ATGTCCCTGGGCAGTTATGTG-3'; 反向引物序列:5'-TGAGAACAGGAACAAGTGGCA- 3',产物大小为512 bp。PCR 反应在ABI 9700 型扩增仪上进行(ABI 公司, 美国), 总体积 20 μL, 其中含1.5 U Taq DNA 聚合酶(GeneAmp High Fidelity Enzyme, 美国ABI 公司)5 U/μL, 2 pmol/L 引物浓度,2.5 mmol/LM dNTP。循环条 件为95℃预变性15 min,先进行95℃变性45 s, 62℃退火或复性45 s(每个循环降0.5℃),72℃ 延伸60 s, 共循环11 次; 然后进行95 ℃ 变性 45 s,57℃退火或复性45 s,72℃延伸60 s,共24 个循环;最后72℃延伸7 min 后降温至4℃备用。 PCR 产物用1% 琼脂糖凝胶电泳检测。 1.2.3 序列测定
应用PCR 产物直接测序的方 法,样品纯化及序列分析由中国优生优育基因科 学专家指导中心应用ABI 3130 XL 型序列分析仪完 成(ABI 公司,美国)。 2 结果
2.1 突变类型 对73 例PKU 患儿共146 个PAH 等位基因外 显子7 及其旁侧内含子区域进行测序分析,结果 检测出6 种突变基因型,分别是R243Q、R241C、 IVS7+2T → A、L255S、G247V 和G247R(图 1), 其等位基因频率分别为14.4%、6.9%、2.7%、0.7%、 0.7% 和0.7%,分为错义突变和剪接位点突变两种 (表1)。73 例患儿中4 例检测出2 个基因突变 (R243Q/R243Q 突变2 例, R243Q/R241C 突变1 例, IVS7+2T → A/G247R 突变1 例),30 例检测出1 个基因突变,外显子7 突变基因总频率为26.0% (38/146),其中回族为28.2%,汉族为23.5% (χ2=1.09,P=0.297)。
 |
图 1 73 例PKU 患儿的6 种PAH 突变基因型 |
| 表 1 73 例PKU 患儿PAH 基因外显子7 突变结果 |
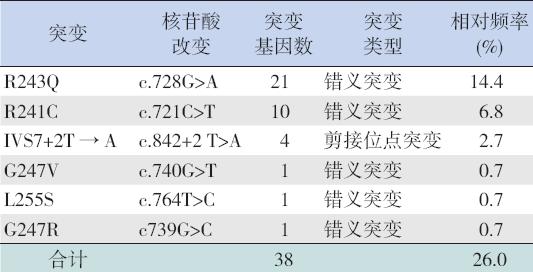
在检出的6 种PAH 突变基因中,突变频率最 高的是R243Q(14.4%),其中回、汉族儿童分别 为14.1%和14.7%(χ2=0.01,P=0.91)。突变频率 居第二位的是R241C(6.8%)。R241C 等位基因 突变的10 例患儿中,有8 例为回族儿童,回族患 儿检出率为10%,高于汉族儿童(3%),差异有 统计学意义(χ2=4.22,P=0.04)。IVS7+2T → A 突 变频率居第三位,G247V、G247R 和L255S 少见。 3 讨论
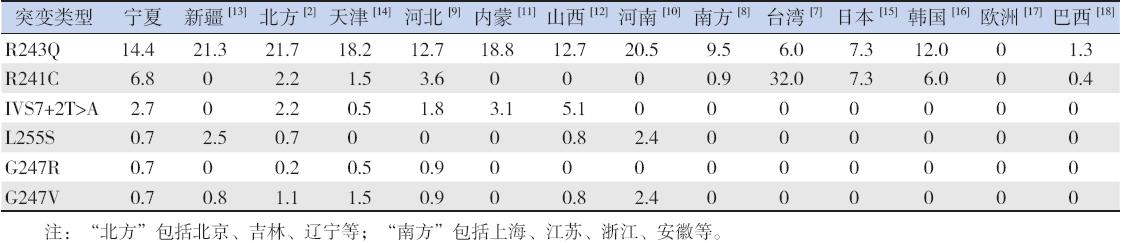
PKU 的病理基础是PAH 基因突变,正常PAH 基因由编码区的13 个外显子和非编码区的12 个 内含子组成,转录1 353 bp 的mRNA,翻译成含 451 个氨基酸的酶单体,PAH 每个亚基的改变都 会引起酶结构和功能的改变,当编码区及其两侧内含子有基因突变发生时,就会引起PAH 的功能 缺失或减低,导致相应的临床表现[5]。PAH 基因 第7 外显子编码PAH 蛋白核心功能区,对保持该 酶活性起关键性作用[6],是基因分析的重点区域。 在13 个外显子中,我国最为常见的PAH 基因突变 区域发生于外显子7,以台湾地区为最高,达到了 42.0%[7],中国南方地区较低,为16.4%[8]。本研究 中外显子7 及其旁侧内含子区域检出率为26.0%, 与河北、河南、内蒙古、山西接近[9, 10, 11, 12],但低于北 方地区[2]、新疆地区[13] 和天津地区[14] 的相关报道。 因此在宁夏地区开展PAH 基因突变研究时应更多 关注外显子7 及其旁侧内含子区域。
本研究在宁夏地区PAH 基因外显子7 中共检 出了6 种突变类型,其中R243Q 等位基因频率最 高(14.4%),但与其他北方地区相比仍属较低频率。 现有报道显示,R243Q 主要存在于亚洲,多集中于 我国北方地区,而在河南的频率达到了20.5%[10]。 处于第二位的R241C 以台湾地区最普遍(32.0%)[7], 日本(7.3%)[15] 和韩国(6.0%)[16] 也较为常见。 但在大陆,宁夏地区R241C 突变比率高于其他地 区。另外4 种突变IVS7+2T → A、L255S、G247R 和G247V 近几年在国内一些地区逐渐被报道, IVS7+2T → A 以山西、内蒙古多见[11, 12],L255S 河南、 新疆多见[10, 13],G247V 河南、天津多见[10, 14]。值得 关注的是G247R 仅散在分布于我国京津冀及宁夏 地区[2, 8, 14],极为少见,似是我国北方地区特有的 一种突变基因类型(表2)。
| 表 2 宁夏地区PAH 基因外显子7 突变型频率与其他地区比较 (%) |
| [1] | 顾学范. 新生儿代谢性疾病筛查[M]. 北京: 人民卫生出版社, 2004: 92-106. |
| [2] | 宋防, 瞿宇晋, 杨艳玲, 等. 中国北方地区苯丙氨酸羟化酶 基因的突变成[J]. 中华医学遗传学杂志, 2007, 24(3): 241-246. |
| [3] | 毛新梅, 马晓燕, 李宏艳, 等. 宁夏回族自治区新生儿疾病 筛查现状调查[J]. 中国妇幼保健, 2012, 27(36): 5988-5990. |
| [4] | Guldberg P, Romano V, Ceratto N, et al. Mutational spectrum of phenylalanine hydroxylase Europe[J]. Hum Mol Genet, 1993, 2(10): 1703-1707. |
| [5] | Scriver CR, Waters PJ, Sarkissian C, et al. PAHab: a locusspecific knowledge base[J]. Hum Mutat, 2000, 15(1): 99-104. |
| [6] | Abadie V, Lyonnet S, Maudfin N, et al. CpG dinucleotides are mutation hot spots in phenylketonuria[J]. Genomics, 1989, 5(4): 936-940. |
| [7] | Chien YH, Chiang SC, Huang A, et al. Mutation spectrum in Taiwanese patients with phenylalanine hydroxylase deficiency and a founder effect for the R241C mutation[J]. Hum Mutat, 2004, 23(2): 206. |
| [8] | 张眉, 顾学范, 张美华, 等. 中国南方人苯丙氨酸羟化酶基 因外显子7 点突变及其频率分析[J]. 中华医学遗传学杂志, 1995, 12(6): 324-326. |
| [9] | 卢超霞, 高峡, 王金玮, 等. 河北地区55 例苯丙酮尿症患者 苯丙氨酸羟化酶基因突变的检测与分析[J]. 中华医学杂志, 2011, 91(42): 2971-2976. |
| [10] | 郭红军, 赵振华, 江淼, 等. 河南地区苯丙酮尿症患者苯丙 氨酸羟化酶基因突变研究[J]. 中华医学遗传学杂志, 2011, 28(2): 142-146. |
| [11] | 张力军, 孟峻, 翟晓萍, 等. 经典型苯丙酮尿症丙氨酸羟化 酶基因的新突变鉴定[J]. 中华医学遗传学杂志, 2005, 22(2): 134-137. |
| [12] | 高伟华, 张全斌, 刘建平, 等. 山西省经典型苯丙酮尿症患 者苯丙氨酸羟化酶基因突变研究[J]. 中华医学遗传学杂志, 2011, 28(4): 393-396. |
| [13] | 余伍忠, 仇东辉, 宋昉, 等. 新疆地区苯丙氨酸羟化酶基因 的突变研究[J]. 中华医学遗传学杂志, 2009, 26(1): 26-30. |
| [14] | 宋力, 党利亨, 孟英韬, 等. 天津及周边地区苯丙氨酸羟化 酶基因突变谱和新突变分析[J]. 中华医学遗传学杂志, 2010, 27(1): 7-12. |
| [15] | Okano Y, Asada M, Kang Y, et al. Molecular characterization of phenylketonuria in Japanese patients[J]. Hum Genet, 1998, 103(5): 613-618. |
| [16] | Lee DH, Koo SK, Lee KS, et al. The molecular basis of phenylketonuria in Koreans[J]. J Hum Genet, 2004, 49(11): 617-621. |
| [17] | Zschocke J. Phenylketonuria mutation in Europe[J]. Hum Mutat, 2003, 21(4): 345-356. |
| [18] | Acosta A, Silva W Jr, Carvalho T, et al. Mutations of the phenylalanine hydroxylase (PAH) gene in Brazilian patients with phenylketonuria[J]. Hum Mutat, 2001, 17(2): 122-130. |