 2014, Vol. 16
2014, Vol. 16

2. 武汉市儿童医院康复科, 湖北 武汉 430016;
3. 武汉市儿童医院神经内科, 湖北 武汉 430016
Rett 综合征(Rett syndrome,RTT)是一种严 重影响儿童精神运动发育的疾病,由 Rett 于 1966 年首次报道,是女性严重智力低下的重要原因; 女性的发病率为 1/10 000~1/15 000 [1] 。1999 年 Amir 等[2] 用定位候选基因的方法确定 RTT 的致病基因 是位于染色体 Xq28 区域的甲基化 CpG 结合蛋白-2 (MECP2)基因,随后研究人员开始对 RTT 进行 MECP2 基因检测,目前世界范围内已经报道了超 过 780 多 种 突 变 类 型(The Human Gene Mutation Database,http://www.hgmd.org)。研究发现大约在 90%~95% 的典型 RTT 及 40%~50% 的非典型 RTT 患者中可检测到 MECP2 基因突变[3],我国总的突 变检出率约为 84%[4] 。本研究通过对 9 例散发 RTT 患儿的临床特点及 MECP2 基因测序分析,了解这 些 RTT 患儿突变情况,为临床诊断在分子水平提 供依据。
1 资料与方法 1.1 研究对象
选取 2012 年至今在本院神经内科和康复科诊 断的 9 例 RTT 患儿为研究对象,均为女孩,发病 年龄 5~42 个月。临床诊断符合 2001 年欧洲小儿 神经会议制定的国际 RTT 诊断标准[5]。本研究得 到武汉市儿童医院伦理委员会的批准。
1.2 基因突变检测
取患儿及其父母外周静脉抗凝血2 mL, 分离白细胞后用酚-氯仿方法提取基因组 DNA。扩增MECP2编码区所需要的引物由上 海生工公司合成,3个外显子序列分别为: 1F:5'-GCTAGGTAAGCTGGGAAATAGC-3', 1R:5'-CACAGTTTGGCACAGTTATGTC-3'; 2F:5'-AAGATCTGAGTGTATGATGGCC-3', 2R:5'-GCACATACATTTTCCTGCTCC-3'; 3F:5'-GTTTGTCAGAGCGTTGTCAC-3',3R: 5'-AAGCTTTGTCAGAGCCCTAC-3'。PCR 总反应体 系为 25 μL,其中含 100 ng DNA 1 μL,50 pmol/L 上 下游引物各1 μL,200 μmol/L dNTP 2 μL,1U Taq DNA 聚合酶(TAKARA 公司 5U/μL)0.2 μL,用无 菌蒸馏水补到 25 μL。在 Bio-rad PCR 仪器上进行 扩增,PCR 条件为 94℃变性 30 s,56℃退火 30 s, 72℃延伸 30 s,循环 32 次。产物经 2% 琼脂糖凝 胶电泳合格后进行测序分析。
2 结果
9 例患儿均具有 RTT 的症状:即语言功能降 低或者丧失;手的技能降低,且部分患儿存在单 一模式手的刻板动作;坐和走的功能受限;多数 患者脑电图显示异常;发育落后于同龄儿童。这 9 例患儿中发现 5 例存在 MECP2 基因突变,1 例 为碱基插入造成的移码突变(c.913insT),该突变 导致 MECP2 基因所编码的蛋白从第 305 位氨基酸 起序列发生改变,且在第330 位氨基酸处提前终止, 该突变未见报道,为新发现突变;2 例为错义突变 (R106W 和 P376S);2 例为无义突变(R168X 和 R270X)。在这些突变中,1 例突变位于外显子 3 上(R106W),其余几个突变均位于外显子 4 上(表 1~表 2)。所有患儿父母均未检测到突变。
|
|
表 1 RTT 患者的临床特点 |

|
|
表 2 RTT 患者 MECP2 基因突变情况 |
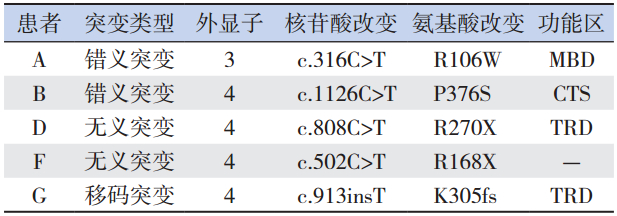
9 例患儿中,1 个突变位于甲基化 CpG 结合 区(MBD),2 个突变位于转录抑制区(TRD), 1 个位于蛋白C 末端区域(CTS),还有1 个突 变位于功能区以外的区域,突变在 MeCP2 蛋白上 的分布如图 1 所示。通过分析发现 2 例(D、G) TRD 功能区有突变的患儿均表现出不说话、脑电 图癎性放电以及发育明显落后的临床特点。
 | 图 1 MECP2 蛋白的结构与功能区划分[6] 及患者 MECP2 基因突变的位置分布 |
3 讨论
RTT 是引起女孩智力低下最常见的原因之一, 多数为散发,临床上分为典型和非典型两类。RTT 患儿一般在 6~18 个月之前表型基本正常,之后会 逐渐表现出智力低下等临床症状[7]。
该病的致病基因 MECP2 编码 X 连锁的甲基化 CpG 结合蛋白-2,是一种高丰度的染色质结合蛋 白[8]。该蛋白共有 486 个氨基酸,包含 2 个主要功 能区:即 MBD 和 TRD[9]。MECP2 不仅可以通过调 节 DNA 甲基化途径影响甲基化过程,还可以作为 转录抑制因子调节基因转录,从而维持与修饰神 经元成熟。因此,如果 MECP2 基因产生突变导致 其编码的蛋白功能异常,可能对神经系统的生长 和发育造成损害[10]。
本研究的 9 例患儿中 5 例 MECP2 基因发生突 变,其中 4 例为 C 突变为 T,与国外报道的 80% 以上的点突变为 C 突变为 T 一致[11]。
患者 A 突变位于 MBD 区域,该区含有 85 个 氨基酸,为蛋白质染色质定位所需[2, 9],该突变可 能改变蛋白的二级结构,导致 MECP2 蛋白与 5-甲 基胞嘧啶的结合能力下降。患者 D 和 G 突变位于 TRD 功能区,该区由 104 个氨基酸组成,它与组 蛋白脱乙酰化酶和转录因子 SIN3A 相互作用发挥 转录抑制作用[2, 9]。患儿 D 的 MECP2 基因突变以 后使得第 270 位氨基酸由精氨酸变为终止密码子; 而患儿 G 的 MECP2 基因外显子 4 插入一个碱基, 产生移码突变使从第 305 位氨基酸(赖氨酸)起 氨基酸序列改变,并在第330 位氨基酸处提前终止, 这 2 例患儿均产生截短的 MECP2 蛋白。这两个突 变可能改变了 TRD 的结构,影响了其转录抑制功 能。患儿 B 突变位于 CTS 区域,推测该区域的突 变影响 TRD 的功能。患者 F 突变虽不在这几个功 能区内,但是该突变使 MECP2 蛋白截短,在 TRD 区域前表达终止,因此 F 患者的 MECP2 蛋白功能 部分或者全部丧失。
有研究对北美国际 Rett 数据库(IRSA,http:// MECP2.chw.edu.au)中的病例统计分析发现超过 80% 的 RTT 患者存在 MECP2 基因突变,剩余的近 20% 突变检测阴性;因此推测可能有其他致病基 因。研究发现一些 RTT 患者中细胞周期依懒性激 酶样 5 蛋白(CDKL5)存在突变[11]。接下来本课 题组将对 MECP2 突变阴性的患者进行 CDKL5 致 病基因的检测。
国内外都有 MECP2 基因突变和 RTT 患儿表 型的研究,包括突变类型以及突变位置的不同对 患者的影响,但是结论不一致[12-13],可能原因是 病例数较少及对临床症状的评分标准不同。本研 究发现位于 TRD 区域的突变表现出不说话、脑电 图癎性放电以及发育明显落后的临床特点,但是 本研究病例较少,不能得出肯定结论,将收集更 多病例进行统计分析,明确基因型和表型的关系。
综上所述,本研究结果和国内外其他研究组 基本一致,MECP2 基因检测可帮助临床进行诊断, 特别是对于临床症状未显现出来的患儿可以尽早 确诊,从而做到早诊断、早干预。
| [1] | Abhishek B, Jorge C, Mriganka S. Rett syndrome: genes, synapasea, circuits, and therapeutics[J]. Front Psychiatry, 2012, 34(3): 1-13. |
| [2] | Amir RE, Van den Vewer IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2[J]. Nat Genet, 1999, 23(2): 185-188. |
| [3] | Weaving LS, Ellaway CJ, Gecz J, et al. RETT syndrome: clinical review and genetic update[J]. J Med Genet, 2005, 42(1): 1-7. |
| [4] | 张晶晶, 包新华, 曹广娜, 等. 中国 Rett综合征患儿突变基因的亲缘分析[J]. 中国医学遗传学杂志, 2010, 27(2): 121-124. |
| [5] | Hagberg B, Hanefeld F, Percy A, et al. An update on clinically applicable diagnostic criteria in Rett syndrome[J]. Eur J Paediatric Neurol, 2002, 6(5): 293-297. |
| [6] | Mimi Wan, Stephen SJ, Zhang XY, et al. Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots[J]. Am J Hum Genet, 1999, 65(6): 1520-1529. |
| [7] | 李颖杰, 朱金玲, 张淑红, 等. Rett综合征的研究进展[J]. 国际遗传学杂志, 2007, 30(3): 229-233. |
| [8] | 潘虹, 王艳萍, 孟洪第, 等. Rett综合征MECP2基因突变分析[J]. 中华医学遗传学杂志, 2002, 19(4): 276-280. |
| [9] | Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin[J]. Cell, 1997, 88(4): 471-481. |
| [10] | Van den Veyver IB, Zoghbi HY. Methyl-CpG-binding protein 2 mutations in Rett syndrome[J]. Curr Opin Genet Dev, 2000, 10(3): 275-279. |
| [11] | Zhang XY, Bao XH, Zhang JJ, et al. Molecular characteristics of Chinese patients with Rett syndrome[J]. Eur J Med Genet, 2012, 55(12): 677-681. |
| [12] | 李美蓉, 潘虹, 包新华, 等. Rett综合征患儿MECP2基因型和表型关系研究[J]. 中华儿科杂志, 2009, 47(2): 124-128. |
| [13] | Schanen C, Houwink EJ, Dorrani N, et al. Phenotypic manifestations of MECP2 mutation in classical an atypic Rett syndrome[J]. Am J Med Genet, 2004, 126(2): 129-140. |