 2014, Vol. 16
2014, Vol. 16



2. 河南省高等学校临床医学重点学科开放实验室, 河南 郑州 450052
噬 血 细 胞 综 合 征(hemophagocytic syndrome, HPS)又称噬血细胞性淋巴组织细胞增生症 (hemophagocytic lymphohistiocytosis,HLH),是由 多种致病因素导致的淋巴细胞、单核细胞和巨噬 细胞系统异常增生活化、以及细胞因子风暴形成 引起的多器官炎症反应综合征。该病临床少见, 病情进展迅猛,病死率高[1]。弥散性血管内凝血 (DIC)是其主要死亡原因之一[2] 。关于影响 HPS 预后的危险因素分析,较一致的结果为血小板计 数明显降低[3, 4, 5, 6]。凝血功能检测结果既受血小板的影响也是早期发现 DIC 的有效指标,其对 HPS 预 后的影响尚未见报道。本文旨在通过分析该病临 床常用指标,寻找影响 HPS 预后的相关危险因素, 为治疗方案的制定提供依据。 1 资料与方法 1.1 一般资料
为排除不同病因对数据分析的影响,本研究 选取 2010 年 1 月至 2012 年 10 月期间我院收治的 35 例感染相关性 HPS 患儿为研究对象,其中男 21 例,女 14 例,男女比例为 1.5 : 1,发病中位年龄为 2 岁(9 个月 ~14 岁),其中 <1 岁 6 例(17%), 1 岁 ~ 14 例(40%),3 岁 ~ 9 例(26%),≥ 7 岁 6 例(17%)。 1.2 诊断及纳入标准
所有患儿均符合国际组织细胞协会 HLH-2004 方案诊断标准[7]。感染相关性HPS 同时满足: (1)无家族性噬血细胞性淋巴组织细胞增生症 (familial hemophagocytic lymphohistiocytosis,FHL) 家族史;(2)相关基因检测无异常;(3)骨髓检查、 淋巴结活检及影像学检查排除恶性疾病;(4)自 身抗体检查排除自身免疫性疾病。 1.3 统计学分析
采用 SPSS 17.0 统计软件对数据进行统计学 分析,符合正态分布的计量资料用均数 ± 标准差 (x±s)表示,两组间比较采用t检验;如不符 合正态分布则用中位数(四分位间距)[M(P25,P75)] 表示,组间比较采用 Wilcoxon 秩和检验。计 数资料用率(%)描述,组间比较用 Fisher 确切概 率法(当n<40 或理论频数 <1 时)。P<0.05 为差 异有统计学意义。 2 结果 2.1 临床表现
35 例患儿均出现发热症状(100%),分别有 31 例(89%)、28 例(80%)和 15 例(43%)患 儿表现出肝脏、脾脏和淋巴结肿大,有呼吸道症 状患儿 18 例(51%),有皮肤出血点患儿 12 例 (34%),表现出黄疸患儿 7 例(20%),消化道 出血 6 例(17%)。 2.2 实验室检查
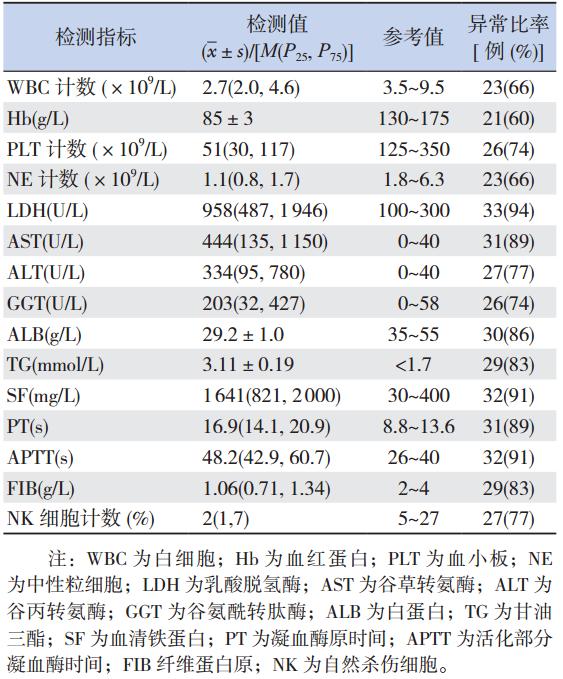
35 例患儿入院时主要实验室检查结果显示: 反应凝血功能的凝血酶原时间(PT)、活化部分 凝血酶时间(APTT)及纤维蛋白原(FIB)等指 标和 PLT 计数以及反应肝功能损伤的乳酸脱氢酶 (LDH)、谷草转氨酶(AST)、谷丙转氨酶(ALT)、 谷氨酰转肽酶(GGT)、白蛋白(ALB)、甘油三 酯(TG)、血清铁蛋白(SF)等指标异常比率均 较高,而粒系和红系减少异常率相对较低,见表 1。
| 表1 35 例 HPS 患儿主要实验室检查结果 |
35 例患儿均进行了骨髓穿刺检查,其中 26 例 (74%)第 1 次骨髓穿刺检查结果以噬血细胞、吞噬红细胞和血小板为主;2 例(6%)第 2 次复查 时发现噬血细胞。7 例(20%)为感染骨髓象。35 例患儿中 31 例(89%)骨髓增生活跃,4 例(11%) 增生低下。 2.4 病原菌分布
35 例患儿中,EB 病毒感染 21 例(60%), 巨细胞病毒感染 13 例(37%),单纯疱疹病毒感 染 9 例(26%),支原体感染 5 例(14%),衣原 体和风疹病毒感染各 4 例(11%),柯萨奇病毒感 染 1 例(3%);有 20 例患儿同时存在两种或两种以上病毒感染。行细菌血培养 31 例,2 例为阳性 结果,其中 1 例为嗜麦芽窄食单胞菌阳性,1 例为 人葡萄球菌阳性;7 例行 GM 试验,2 例阳性。 2.5 治疗及转归
35 例 病 例 中,25 例 严 格 按 HLH-2004 方 案[2] 治疗,其中 3 例死亡,22 例存活,治疗有效 率 88%;10 例除未使用依托泊甙(VP-16)外,其 余治疗方案均按 HLH-2004 方案进行,其中 5 例死 亡,5 例存活,治疗有效率 50%。两种治疗方案比 较,疗效差异有统计学意义(P=0.027)。8 例死 亡病例均于入院早期死亡,中位死亡时间为 4 d(1~ 52 d)。 2.6 不同结局患儿主要指标比较
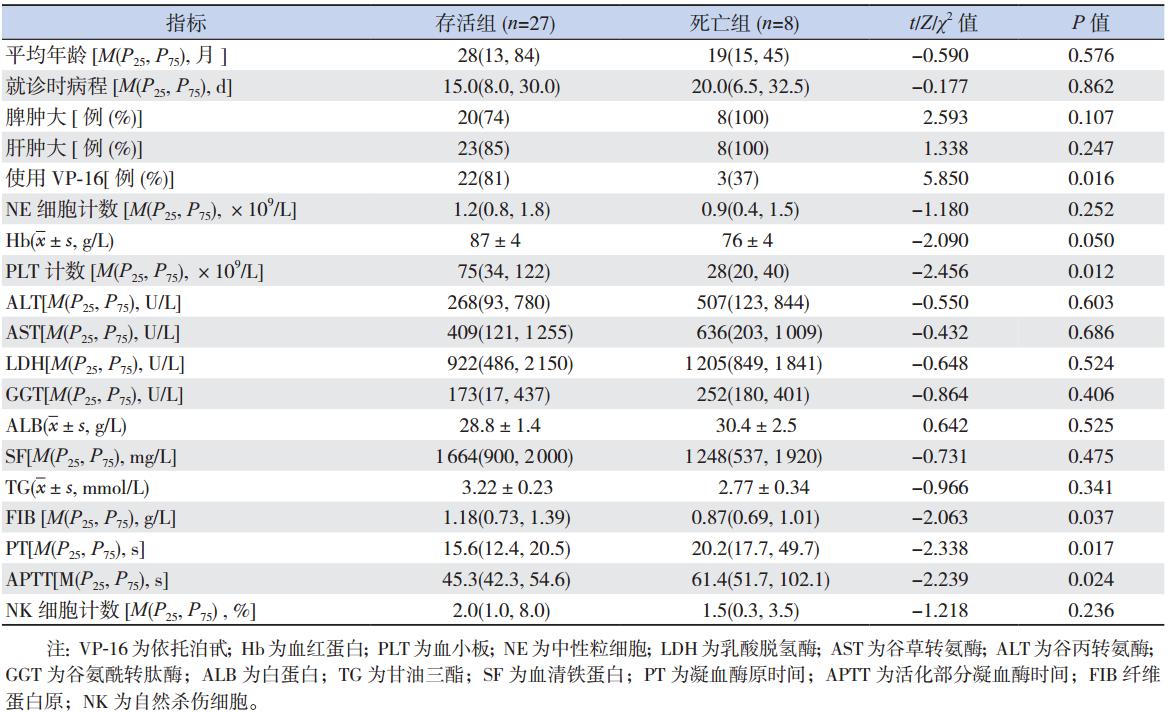
死亡病例与存活病例主要指标比较显示:未 使用 VP-16 治疗、就诊时即出现 PLT 计数减低及 PT、APTT、FIB 等指标异常者预后不良,见表 2。
| 表 2 不同结局患儿主要指标比较 |
HPS 分为原发性和继发性。原发性 HPS 为常 染色体隐性遗传。但基因缺陷仅是 HPS 发病的基 础背景,单独的基因缺陷并不足以导致 HPS 发病, 往往由继发因素包括感染、肿瘤、免疫缺陷病及 组织损伤等诱发[8]。继发性 HPS 病因主要有感染、 恶性肿瘤、自身免疫性疾病等。不管原发性或继 发性 HPS,感染都是最常见的病因,而感染的病 例又以 EB 病毒感染最多[9]。本研究35例HPS患 儿中 EBV 感染者有 21 例(21/35,60%),与上 述报道[9] 一致。
HPS 病情进展迅猛,病死率高。大部分研究 结果显示严重肝功能损伤、PLT 计数降低、低 FIB 血症等指标是影响 HPS 预后的危险因素[3, 4, 5, 6]。本研 究结果显示在疾病早期死亡组和存活组比较,年 龄、就诊时病程、AST、ALT、GGT、LDH、TG、 SF、NK 细胞计数等指标无显著差异,而 PLT 计数、 PT、APTT 及 FIB 等指标有显著差异。分析上述 指标发现这些因素都会影响到凝血功能,而凝血 功能障碍引发的 DIC 也是 HPS 的主要死亡原因之 一[7]。国内孙雪辉等[10] 对 11 例 HPS 患者临床特点 进行分析后,发现所有并发 DIC 的患者均死亡。本 研究也显示 8 例死亡病例均因早期并发 DIC 死亡。 因此,早期发现凝血异常,及时纠正,预防 DIC 的 发生可能成为降低本病病死率的重要治疗手段。
HPS 并发 DIC 的机制主要有以下三个方面: (1)PLT 计数减少:PLT 降低的程度与 HPS 预后 密切相关[11]。HPS 患儿 PLT 减少的主要原因为高 水平细胞因子直接抑制骨髓造血。另外,异常增殖、 活化的巨噬细胞吞噬和引发 DIC 后的消耗都造成 了 PLT 计数减低。(2)肝功能损害[9]:肝脏是合 成凝血因子的重要场所,除Ⅲ、Ⅳ、Ⅴ外的所有 凝血因子均在肝脏合成,此外肝脏还合成多种抗 凝血因子和纤溶系统化合物等,因此肝脏在体内 的止凝血过程中起着非常重要的作用。(3)严重 感染:感染时机体产生大量的 IL-1、TNF 等前炎 性细胞因子,直接损伤血管内皮细胞,使内皮下 胶原暴露,从而启动内源性凝血系统;同时损伤 的血管内皮细胞释放的组织因子结合并活化因子 Ⅶ,激活外源性凝血系统,是 DIC 发病的重要机制。
针对以上病因,传统的 DIC 治疗一般包括肝 素抗凝及补充 PLT、凝血因子、FIB 等对症支持治 疗的同时尽快控制原发病,去除诱因。对于 HPS 诱发的 DIC 治疗的关键在于减少炎症因子风暴对 各个脏器的损害,尽早控制 HPS 的发展。血浆置 换法也是快速去除血清中高细胞因子可靠和有效 的治疗方法[12]。另外,Zhuang 等[13] 报道使用重组 人Ⅶ a 因子可快速纠正 DIC,减少出血并为治疗原 发病争取时间。最近有研究报道了 1 例使用重组 人血栓调节蛋白(r-TM)+VP-16 治疗 HPS 诱发的 DIC [14] 。r-TM 作为一种新的抗凝剂,临床试验显示 疗效优于肝素,在日本被广泛用于 DIC 治疗。 本研究中使用 VP-16 和未使用 VP-16 的病例 比较,使用VP-16 治疗有效率明显高于未使用患儿。 VP-16 为细胞凋亡的启动剂,对单核细胞性和组织 细胞性疾病具有高度活性,通过诱导异常增殖的 组织细胞凋亡达到降低 HPS 患者血清细胞因子水 平的作用。VP-16 于 1980 年开始用于 HPS 的治疗, 取得了较好的疗效,在 HLH-2004 方案中将其提前 到了治疗开始,即患者 WBC 及 PLT 计数无明显降 低的情况下就开始应用[15]。国内李小琳等[16] 研究 结果也显示未使用 VP-16 或诊断后 4 周再使用, 患儿死亡风险大大增加,故建议早期应用。
综上,本研究结果显示 DIC 仍是 HPS 患儿死 亡的主要原因,但一直以来对于并发 DIC 后的治 疗并没有新的突破,那么及早发现并预防 DIC 的发生便成为降低 HPS 病死率的重要治疗手段。本 研究结果提示可将凝血功能监测作为常规检查手 段,积极纠正凝血异常,严格按照 HLH-2004 方案 治疗 HPS,将对改善 HPS 临床预后发挥重要作用。
| [1] | Bhattacharyya M, Ghosh MK. Hemophagoctic lymphohistiocytosis-recent concept[J]. J Assoc Physicians India, 2008, 56: 453-457. |
| [2] | 闫丽娟, 王昭. 噬血细胞综合征继发凝血功能障碍的认识与研究[J]. 临床和实验医学杂志, 2012, 11(2): 142-143. |
| [3] | 肖莉, 宪莹, 戴碧涛, 等. HLH-2004方案治疗83例EB病毒相关噬血淋巴组织细胞增生症患儿疗效分析[J]. 中华血液学杂志, 2011, 32(10): 668-672. |
| [4] | 王华, 高文瑾, 刘安生, 等. 儿童噬血细胞综合征54例临床及预后因素分析[J]. 中国小儿血液与肿瘤杂志, 2013, 18(1): 31-34. |
| [5] | Tseng YT, Sheng WH, Lin BH, et al. Causes, clinical symptoms,and outcomes of infectious disease associated with hemophagocytic lymphohistiocytosis in Taiwanese adults[J]. Microbiol Immunol Infect, 2011, 44(3): 191-197. |
| [6] | Karapinar B, Yilmaz D, Balkan C. An unusual cause of multiple organ dysfunction syndrome in the pediatric intensive care unit : hemophagocytic lymphohistiocytosis[J]. Pediatr Crit Care Med, 2009, 10(3): 285-290. |
| [7] | 中华医学会儿科学分会血液学组. 噬血细胞性淋巴组织细胞增生症诊疗建议[J]. 中华儿科杂志, 2012, 50(11): 821-825. |
| [8] | 汤静燕, 李志光. 儿童肿瘤诊断与治疗学[M]. 北京: 人民军医出版社, 2011: 282-289. |
| [9] | Ansuini V, Donato R, Esposito S. Debate around infection-dependent hemophayocytic syndrome in paediatrics[J]. BMC Infection Diseases, 2013, 16(1): 13-15. |
| [10] | 孙雪辉, 郑文浩, 张文, 等. 自身免疫病合并噬血细胞综合征临床分析[J]. 中华内科杂志, 2010, 49(10): 836-840. |
| [11] | 王旖旎, 王昭, 吴林. 多中心72例噬血细胞综合征诊疗分析[J]. 中华血液学杂志, 2009, 30(12): 793-798. |
| [12] | 郭子雄, 钟敏泉, 翟琼香, 等. 小儿噬血细胞综合征合并多脏器功能不全临床特征研究[J].中华临床医师杂志, 2011, 5(8): 2374-2377. |
| [13] | Zhuang JL, Jiang QW, Xu Y, et al. Recombinant activated factor Ⅶ in hemophagocytic lymphohistiocytosis with disseminated intravascular coagulation[J]. Chin Med J(Engl), 2011, 124(19): 3189-3191. |
| [14] | Yamamoto M, Hori T, Hatakeyama N, et al. Use of recombinant thrombomodulin in disseminated intravascular cogulation complicated hemophagocytic lymphohistiocytosis[J]. Indian J Pediatr, 2014, 81(3): 288-291. |
| [15] | Janka GE. Hemophagocytic lymphohistiocytosis[J]. Blood Rev, 2007, 21(5): 245-253. |
| [16] | 李小琳, 刘玉玲, 付四毛. 儿童噬血细胞综合征32例临床分析[J]. 中国小儿急救医学, 2012, 9(1): 38-40. |