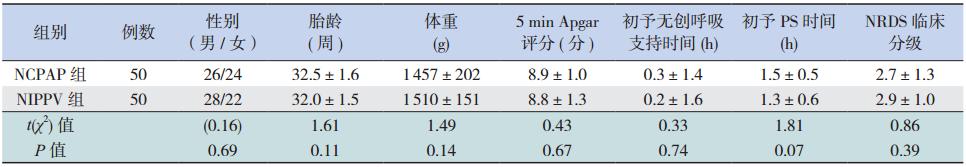
 2014, Vol. 16
2014, Vol. 16



X-连锁低血磷抗D佝偻病(X-linked hypophosphatemic rickets,XLH)是一组由于肾小管 磷酸盐转运异常和重吸收障碍,导致大量尿磷、 低血磷、骨质矿化缺乏和维生素 D 代谢异常的显 性遗传性疾病[1, 2, 3] 。其发病率为 1/20 000,是遗传 性佝偻病中最常见的一型[4, 5]。本病的临床表现在儿童主要为生长发育迟缓,身材矮小,骨骼疼痛, 行走无力,双下肢弯曲畸形,骨质疏松,多发性 骨折以及牙釉质发育不良[6];在成人,主要为软骨 病和骨关节畸形[7]。实验室检查可发现患者的血磷 明显降低,血钙正常,甲状旁腺激素正常或增高 以及血 1,25-羟维生素 D3正常或轻度下降[8]。
HYP Consortium 于 1995 年证明磷酸盐调节 基因 (PHEX)为此病的致病基因[9]。此基因 (OMIM300550)定位于染色体 XP22.1-22.2,全 长 243 kb,由 22 个外显子构成,其编码的跨膜蛋 白含 729 个氨基酸,属于二型锌依赖性内肽酶家 族整合蛋白的成员。研究证实人类和小鼠的 PHEX 序列有 96% 的同源性[10]。自 1986 年刘向善[11] 报 道了中国第 1 例低磷抗维生素 D 佝偻病以来,国 内对该病的研究多为病例报道,分子遗传学研究 报道数量较少。本研究报道国内 3 例基因诊断明 确的 XLH,并复习以往相关文献,探讨中国 XLH 患者存在的突变热点和突变类型,以提高对该疾 病的认识。 1 资料与方法 1.1 研究对象
研究对象为我院2011年11月至2012年6月 疑诊为 XLH 的 3 例患儿,其中男 1 例,女 2 例, 年龄 3~9 岁,1 例有家族史(母亲患病),2 例为 散发病例。 1.2 诊断标准
由于国内外对于低磷抗维生素 D 佝偻病没
有严格统一的标准,参考诸福棠实用儿科学[12],
XLH 的临床特点为:于生后第 1 年出现典型的双
下肢弓形腿,伴身高增长慢,无手足抽搐;在儿
童期,常有颅缝晚闭,四肢大关节可发生致畸性
骨痛关节炎,可有出牙晚,牙痛,牙齿易脱落;
血清钙正常,血清磷明显降低合并尿磷增高(肾
小管磷重吸收率降低),碱性磷酸酶升高,甲状
旁腺功能可正常或轻度升高,双膝放射线检查可
有典型佝偻病改变。回顾性分析了 3 例患儿的临
床资料,患儿均有身材矮小、佝偻病、低磷血症、
尿磷增高等表现,符合 XLH 的临床特点(表 1)。
取得患儿监护人知情同意后,留取患儿及其
父母外周静脉血 2 mL(肝素抗凝),用博迈德全
血基因组提取试剂盒提取基因组 DNA,测定浓度
后 -20℃留存备用。
1.4 PCR 扩增
通过 UCSC 数据库(http://genome.ucsc.edu/)
得 到 PHEX 基 因 序 列(NM_000444),应 用
Primer 3.0 设计引物,对 PHEX 基因的 22 个编码
外显子及其相邻的内含子序列进行 PCR 扩增。
在 20 μL 反应体系中,含 10×PCR 缓冲液 2 μL,
2.5 mmol/L 的 dNTP 3 μL,5 U/μL 的 rTaq 酶 0.3 μL
大连宝生物有限公司),100 ng/μL 基因组 DNA
1 μL,正反向引物各1 μL,去离子水 11.7 μL。PCR 反应条件为:95℃ 预变性 5 min,94℃变性
30 s,59℃退火 30 s,72℃延伸 45 s,共 35 个循环,
最后 72℃延伸 5 min。PCR 产物送北京天一辉远公
司切胶纯化和基因测序。
1.5 突变的证实
针对所发现的突变首先进行反向测序,结果
与 UCSC 数据库基因组序列进行比对分析,进一步
检测患儿父母外周血 DNA,了解是否也能发现相
同位点的突变。
2 结果
2.1 临床特点
3 例患儿中,1 例男性患儿以前额突出就诊,
2 例女性患儿以双下肢弯曲就诊。起病年龄为 5 个
月至 2 岁,诊断年龄为 2~3 岁。就诊时患儿身高
均小于 -3 SD,且均有膝内翻、步态不稳和方颅。
肋骨串珠 1 例,肋缘外翻 2 例,牙齿脱落 1 例。
3 例病例血清磷明显降低(0.70~0.86 mmol/L),
24 h 尿磷明显增高(1.05~1.43 mmol/kg)。血碱性
磷酸酶升高(490~949 U/L)。1 例患儿血甲状旁
腺激素升高(109 pg/mL)。3 例患儿双上肢 + 下
肢 X 片显示符合佝偻病征象(双侧肱骨、尺桡骨、
胫腓骨骨干弯曲,骨质密度减低,干骺端呈杯口状,
毛刷样改变,干骺端发育延迟)(图 1)。病例 3
在 1.5 岁初诊时外院曾误诊为维生素 D 缺乏性佝偻
病,此后手术矫正 O 型腿 3 次。
将 PHEX 的 22 个编码外显子的测序结果与
正常序列比对,发现 3 例病例均存在基因突变,
包括无义突变和剪接突变 2 种类型。其中已报道
的突变有 2 例:外显子 1 上碱基序列 58 处的胞
嘧啶由胸腺嘧啶代替,从而导致终止密码的产生
(c.58C>T,p.R20X)及在第 15 外显子和内含子
连接处发生单碱基置换(c.1645+1G>A)。新发现
的突变 1 例,为第 4 内含子起始处的剪接位点发
生的改变(c.436+1G>A)。病例 1、3 的父母均未
检测到突变,病例 2 和其母亲均检测相同突变,
其父亲未检测到突变(图 2、3)。
至 2014 年 1 月,世界上已报道的 XLH 患者
PHEX 基因突变共有 329 种,覆盖了 PHEX 基因的全长(http://www.phexdb.mcgill.ca/)。其中最常见
的为错义突变(24%),突变热点区域有 3 个:外
显子 8~10,外显子 14~17 和外显子 19~22[13]。不
同地区的病例总数和突变类型存在差异,欧洲 267
例报道中最常见的突变为框移突变(24%);美
洲 10 例报道中最常见的为错义突变(40%);亚
洲 51 例报道中最常见的为无义突变(31%)[14]。
经 过 检 索 Pubmed(http://www.ncbi.nlm.nih.gov/
pubmed)和万方(http://www.wanfangdata.com.cn/)
数据库,共检索到 12 篇关于中国 XLH 患者进行
PHEX 基因检测的报道。12 篇文献共报道 89 例
XLH 病人,发现 28 种突变,其中在 22 外显子上
发现的突变数量最多(18%),突变类型最多的
为错义突变(61%)(表 2)。文献曾报道在欧洲
地区 XLH 患者中,散发病例占病人总数的 40%,
而 PHEX 基因突变检出率在有家族史的病人中是
87%,在散发病例中是 73%。中国已报道的 89 例
XLH 病人中,28 例为散发病例,PHEX 基因突变
检出率为 100%,61 例有家族史,PHEX 基因突变
检出率为 88%(54/61)[3, 7, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22]。本组资料的3 例
患儿中,1 例有家族史,2 例为散发病例,均检出
PHEX 基因突变。
病例 2 检出的无义突变(c.58C>T,p.R20X)
位于外显子 1 上,处在 PHEX 编码的跨膜蛋白 N
端的胞质侧,使 mRNA 第 20 位的精氨酸处终止转
录,从而造成蛋白的截短。Dixon 等[23] 于 1998 年
曾预测大部分(70%)发生在 PHEX 基因上的突
变会导致其蛋白功能的丧失,而并不是由于单倍
剂量不足或者显性负效所导致。迄今为止,世界
上发现此突变的报道共有 7 例,病例人群覆盖的
地域包括了俄罗斯、芬兰、波兰、德国、比利时
和韩国,在中国人群中还未见报道[4, 23, 24, 25, 26, 27, 28]。2000 年
Tyynismaa 等
[26] 曾推测此突变没有人种特异性,为
常见的突变之一。在病例 1 和其母亲检测出的剪
接突变(c.1645+1G>A)位于 15 外显子和内含子
的 5’剪接供体位置处。最初于 2001 年在澳大利
亚被报道,迄今共报道 8 例患者[2, 29, 30, 31]。虽然此突
变所导致的 RNA 剪接效应和蛋白功能研究尚不足,
此位点在恒河猴、小鼠、狗等物种中都极度保守,
且此处 5’端的剪接强度根据 Neural Network 剪接
预测软件(www.fruitfly.org/seq_tools/splice.html)分
析后,发现此处为 G 的剪接强度为 0.96(正常值
0~1),变为 A 后剪接信号极为微弱(<0),且根
据数据分析,此处的突变也可能影响 15 内含子的
3’剪接受体,从而导致外显子跳跃。而病例 3 检
测出的剪接突变(c.436+1G>A)为一例新突变。
在内含子 4 上剪接位点的突变共有 4 例报道,分
别为 c.436+4A>C,c.436+3G>C,c.436+6C>T 和 437-3C>G
[29, 32, 33]
。除了荷兰在 2008 年报道的 c.436+6C>T
从 RNA 水平上证实了此剪接突变导致了外显子 4
的跳跃剪接外,余报道均未做相关功能的研究[33]。
本研究报道的突变经过 Neural Network 预测后发现
此处 5’端 G 的剪接强度为 0.92,而变为 A 后强
度为 0.1,所以预测此突变会导致错误剪接。
目前,PHEX 基因的基因型和表型没有发现联系[14]。XLH 患者的临床表现多样,且严重程度
差异很大,主要是靠家族史和临床表现诊断。此
病在儿童期主要表现为生长发育迟缓,包括双下
肢弯曲、出牙延迟和方颅[6]
。Drezer
[6]在2000 年分析,
XLH 患者的大部分症状在6~12 个月之后才会出现,
而且对于没有家族史的患者,80% 在 2~3 岁以后
因为双下肢弯曲而就诊。在中国已报道的 89 例
病人中,可获取资料的为 42 例,其中小于 18 岁
的 22 例,平均诊断年龄为 6 岁。所有患者就诊原
因均为双腿弯曲,其中有 20 例发现方颅,且起病
平均从 6 个月开始,首发症状均为生长迟缓和方
颅[3, 13, 16, 19]。本组资料中的 3 例患者诊断平均年龄
为 5 岁,均发现方颅。病例 1 从 5 个月开始出现
前额突出,家长未予重视,病例 2 和 3 出现方颅
的时间不详。因此推测 1 岁以内 XLH 的患儿可以
通过方颅等早期佝偻病的体征及时诊断,从而进
行早期治疗。根据文献报道,XLH 病例的骨骼畸
形随年龄增长而逐渐加重,早期及时的诊断和长
期的治疗是决定患者预后的关键[6, 34]。
本研究中,病例 3 曾在 1.5 岁由外院误诊为维
生素 D 缺乏性佝偻病。而国内也有多篇文献分析
了 XLH 患者的误诊,主要被误诊的疾病皆为维生
素 D 缺乏性佝偻病[34, 35]。XLH 是一种以低磷血症
为特征导致骨发育和营养不良的遗传性骨病,有
阳性的家族史,与维生素 D 缺乏性佝偻病相比,
主要特点为:血磷降低显著,尿磷增高,血钙正
常或稍低,罕有抽搐。佝偻病在 2 岁以后仍有活动,
病情持续进展;而后者血磷正常,血钙降低显著,
严重者出现婴儿手足搐搦症。1,25-(OH)D3 下降明
显。佝偻病在 2 岁后进入后遗症期,没有活动性,
病情不进展。XLH 对于常规剂量维生素 D 和钙剂
治疗无效,本病需磷酸盐合剂和大剂量维生素 D
同时治疗方可有效,以上几点可作为鉴别诊断的
主要方法。另外,佝偻病方面 XLH 还需要和原发
性肾小管酸中毒、范可尼综合征等鉴别。
PHEX 基因作为 XLH 的致病基因,几乎所有
的 PHEX 基因突变均会使 PHEX 在成骨细胞表面
的功能受损。PHEX 在骨骼、牙齿、脑、肌肉、睾
丸中均有表达,但在肾脏中无表达,说明 PHEX
蛋白异常不是直接作用于肾脏来影响磷的回收和
维生素 D 的代谢[36]。Shimada 等[37]
和 White 等 [38]
多次在实验中证实 FGF-23 作为重要的调磷因子,是 PHEX 蛋白的分解底物。当 XLH 患者基因突变
时,其作用底物堆积,FGF-23 的浓度在患者体内
显著升高,从而影响肾小管对磷的重吸收。2012
年美国内分泌协会将 XLH 与常染色体显性遗传性
低磷性佝偻病(ADHR)、常染色体隐性遗传性佝
偻病(ARHR)、肿瘤诱发的骨软化症(TIO)等
8 种疾病分类于 FGF-23 介导的低磷血症
[39]。可以
说 FGF-23 的检测对于 XLH 的患者具有重大意义。
由于技术所限,本研究并未检测患儿体内的 FGF-23 水平,以后应当开展此部分的工作,以对 XLH
进行更加深入的探索。
综上所述,本研究从临床和基因水平上确诊
了 3 例 XLH 病例,此 3 例患儿 PHEX 基因突变在
中国人群中均是首次报道,在国际上其中 2 例为
已报突变,1 例为新突变。另外,本研究还对相
关文献进行了复习和总结,发现中国 XLH 病人在
PHEX 基因报道中最常见的突变位置为第 22 外显
子,最常见的突变类型为错义突变。今后,我们
需要对中国 XLH 的病例进行更大样本的研究,以
明确最常见的突变热点,以及在分子遗传学上面
对基因型和表型的联系进行进一步的探讨。
![]()
表1 12 3 例患儿的临床资料

图 1 病例 1 的影像学表现 尺桡骨 X 片显示:骨干弯
曲,骨质密度减低,干骺端呈杯口状,毛刷样改变,干骺端发育
延迟。

图 2 病例 1、3 的测序结果 A:病例 1 测序图,显
示在第15 外显子和内含子连接处发生单碱基置换(c.1645+1G>A);
B: 病例 3 测序图,显示第 4 内含子起始处的剪接位点发生的改变
(c.436+1G>A)。此 2 例突变均为剪接突变。箭头所示为突变位点。

图 3 病例 2 及其母亲的测序结果 A:病例 2 测序图,
显示在第 1 外显子上第 58 处的胞嘧啶由胸腺嘧啶代替,从而导致
无义突变(c.58C>T,p.R20X);B:病例 2 的母亲测序图,显示
在相同位点发现同样突变。箭头所示为突变位点。
![]()
表 2 中国人群 PHEX 基因已知突变
| [1] | Hanna JD, Niimi K, Chan JM. X-linked hypophosphatemia. Genetic and clinical correlates[J]. Am J Dis Child, 1991, 145(8): 865-870. |
| [2] | Holm IA, Nelson AE, Robinson BG, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets[J]. J Clin Endocrinol Metab, 2001, 86(8): 3889-3899. |
| [3] | Kang Q, Xu J, Zhang Z, et al. Three novel PHEX gene mutations in four Chinese families with X-linked dominant hypophosphatemic rickets[J]. Biochem Biophy Res Commun, 2012, 423(4): 793-798. |
| [4] | Francis F, Strom TM, Hennig S, et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets[J]. Genome Res, 1997, 7(6): 573-585. |
| [5] | Shows TB, McAlpine PJ. The catalog of human genes and chromosome assignments. A report on human genetic nomenclature and genes that have been mapped in man[J]. Cytogenet Cell Genet, 1978, 22(1-6): 132-145. |
| [6] | Drezner MK. PHEX gene and hypophosphatemia[J]. Kidney Int, 2000, 57(1): 9-18. |
| [7] | 王静,金春莲,任梅宏,等. 抗维生素D佝偻病PHEX基因突变分析[J]. 国际遗传学杂志, 2008, 31(4): 251-254. |
| [8] | Brame LA, White KE, Econs MJ. Renal phosphate wasting disorders: clinical features and pathogenesis[J]. Semin Nephrol, 2004, 24(1): 39-47. |
| [9] | Francis F, Hennig S, Korn B, et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium[J]. Nat Genet, 1995, 11(2): 130-136. |
| [10] | Guo R, Quarles LD. Cloning and sequencing of human PEX from a bone cDNA library: evidence for its developmental stage-specific regulation in osteoblasts[J]. J Bone Miner Res, 1997. 12(7): 1009-1017. |
| [11] | 刘向善. 低磷抗维生素D性佝偻病一家系4例报告[J]. 实用内科杂志.1986, 7(1): 56. |
| [12] | 倪佳臣.肾小管运转功能障碍[M]//胡亚美,江载芳. 诸福棠实用儿科学下册. 第7版. 北京:人民卫生出版社,2002: 2163-2164. |
| [13] | 曹丽华,麻宏伟,王述森,等. 低磷酸盐血症性佝偻病PHEX基因突变分析[J]. 国际遗传学杂志, 2011, 34(3): 123-126. |
| [14] | 宋莹, 麻宏伟, 黎芳, 等. X-连锁低磷性佝偻病的基因突变分析[J]. 中国当代儿科杂志, 2013, 15 (11): 928-931. |
| [15] | Chou YY, Chao SC, Tsai SC, et al. Novel PHEX gene mutations in two Taiwanese patients with hypophosphatemic rickets[J]. J Formos Med Assoc, 2005, 104(3): 198-202. |
| [16] | Lo FS, Kuo MT, Wang CJ, et al. Two novel PHEX mutations in Taiwanese patients with X-linked hypophosphatemic rickets[J]. Nephron Physiol, 2006, 103(4): 157-163. |
| [17] | Xia W, Meng X, Jiang Y, et al. Three novel mutations of the PHEX gene in three Chinese families with X-linked dominant hypophosphatemic rickets[J]. Calcif Tissue Int, 2007, 81(6): 415-420. |
| [18] | Qiu G, Liu C, Zhou J, et al. Prenatal diagnosis for a novel splice mutation of PHEX gene in a large Han Chinese family affected with X-linked hypophosphatemic rickets[J]. Genet Test Mol Biomarkers, 2010, 14(3): 385-391. |
| [19] | Jap TS, Chiu CY, Niu DM, et al. Three novel mutations in the PHEX gene in Chinese subjects with hypophosphatemic rickets extends genotypic variability[J]. Calcif Tissue Int, 2011, 88(5): 370-377. |
| [20] | 唐佳,潘敬新,蒋玮莹,等. 一例抗维生素D佝偻病的基因诊断和新突变的致病性鉴定[J]. 中华临床医师杂志, 2012, 6(5): 1226-1230. |
| [21] | Yang L, Yang J, Huang X. PHEX gene mutation in a Chinese family with six cases of X-linked hypophosphatemic rickets[J]. J Pediatr Endocrinol Metab, 2013, 26(11-12): 1179-1183. |
| [22] | 马明义, 李华, 蔡延森. 一个低血磷性佝偻病家系的 PHEX 基因突变研究[J]. 中华医学遗传学杂志, 2013, 30(5): 582-584. |
| [23] | Dixon PH, Christie PT. Mutational analysis of PHEX gene in X-linked hypophosphatemia[J]. J Clin Endocrinol Metab, 1998, 83(10): 3615-3623. |
| [24] | Cho HY, Lee BH, Kang JH, et al. A clinical and molecular genetic study of hypophosphatemic rickets in children [J]. Pediatr Res, 2005, 58(2): 329-333. |
| [25] | Rowe PSN, Oudet CL, Francis F, et al. Distribution of mutations in the PEX gene in families with X-linked hypophosphataemic rickets (HYP) [J]. Hum Mol Genet, 1997, 6(4): 539-549. |
| [26] | Tyynismaa H, Kaitila I, Nanto-Salonen K, et al. Identification of fifteen novel PHEX gene mutations in Finnish patients with hypophosphatemic rickets [J]. Hum Mutat, 2000, 15(4): 383-384. |
| [27] | Popowska E, Pronicka E, Sulek A, et al. X-linked hypophosphatemia in Polish patients. 1. Mutations in the PHEX gene [J]. J Appl Genet, 2000, 41(4): 293-302. |
| [28] | Popowska E, Pronicka E, Sulek A, et al. X-linked hypophosphatemia in Polish patients. 2. Analysis of clinical features and genotype-phenotype correlation [J]. J Appl Genet, 2001, 42(1): 73-88. |
| [29] | Gaucher C, Walrant-Debray O, Nguyen TM, et al. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets [J]. Hum Genet, 2009, 125(4): 401-411. |
| [30] | Beck-Nielsen SS, Brixen K, Gram J, et al. Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and CLCN5 in patients with hypophosphatemic rickets [J]. J Hum Genet, 2012, 57(7): 453-458. |
| [31] | Durmaz E, Zou M, Al-Rijjal RA, et al. Novel and de novo PHEX mutations in patients with hypophosphatemic rickets [J]. Bone, 2013, 52(1): 286-291. |
| [32] | Filisetti D, Ostermann G, von Bredow M, et al. Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues [J]. Eur J Hum Genet, 1999, 7(5): 615-619. |
| [33] | Makras P, Hamdy NAT, Kant SG, et al. Normal growth and muscle dysfunction in X-linked hypophosphatemic rickets associated with a novel mutation in the PHEX gene[J]. J Clin Endocrinol Metab, 2008, 93(4): 1386-1389. |
| [34] | 刘丽君, 郝淑青, 刘玲, 等. 家族性低磷抗D佝偻病六例误诊分析[J]. 中国全科医学, 2004, 7(2): 1434-1435. |
| [35] | 吕娜, 魏素芳, 乔玉巧, 等. 儿童低磷抗维生素D佝偻病23例[J]. 临床荟萃, 2008, 23(3): 205-206. |
| [36] | 夏维波,孟迅吾,周学瀛. 调磷因子:成纤维细胞生长因子23[J]. 国外医学内分泌学分册, 2004, 24(6): 241-243. |
| [37] | Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis[J]. J Bone Miner Res, 2004, 19(3): 429-435. |
| [38] | White KE, Carn G, Lorenz-Depiereux B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23[J]. Kidney Int, 2001, 60(6): 2079-2086. |
| [39] | Imel EA, Econs MJ. Approach to the hypophosphatemic patient. [J]. J Clin Endocrinol Metab, 2012, 97(3): 696-706. |