 2014, Vol. 16
2014, Vol. 16



Menkes 病(MIM#309400) 是一种罕见的X 连锁隐性遗传病,由于ATP7A 基因突变导致小肠 上皮细胞铜转运机制障碍,体内铜缺乏,铜相关 酶的功能缺陷引起多系统功能障碍。自1962 年 Menkes 等[1] 首次报道以来,至今全球已报道了 近400 例病例。据文献报道欧洲Menkes 病发病率 约为1/300 000,日本约为1/360 000[2],我国关于 Menkes 病的研究较少[3, 4, 5],缺乏流行病学研究资料。 Menkes 病的主要临床表现为神经系统损害、结缔 组织异常、结节性脆发。男性患者通常在生后数 月发病,多于早期儿童期死亡。Menkes 病的诊断 主要依据典型的临床表现,特别是毛发改变,血 清铜和血浆铜蓝蛋白检测是关键的生化诊断线索。 X 片、头颅MRI、动脉造影亦有助于鉴别诊断。 ATP7A 基因检测是确诊Menkes 病的关键技术[6], 迄今国内外已发现约300 种突变[7]。皮下注射组氨 酸铜是最有效的治疗,可显著改善患者预后[8],但 是,由于缺乏合法的药物来源,患者难以获得治疗。 本文就2012 年4 月至2013 年1 月我院诊断的3 例Menkes 病患儿的临床经过、生化表现和基因突 变进行研究,探讨Menkes 病的临床特点、诊断与 产前诊断方法,提高认识。 1 资料与方法 1.1 研究对象
3 例患儿均为男婴,2012 年4 月至2013 年1 月于我院儿科就诊,于8~9 个月龄获得诊断。3 例 患儿分别于生后2、4、5 个月时起病,主要临床 表现为抽搐和智力运动落后。患儿父母均为非近 亲结婚,身体健康。仅1 例患儿(例1)有异常家 族史,其母亲第1 胎妊娠4 个月时发现胎儿脊柱裂, 引产。 1.2 方法
对3 例患儿进行了详细临床调查、一般化验、 血清铜蓝蛋白检测、脑电图、脑磁共振扫描。
ATP7A 基因检测:在患儿家属知情同意的前 提下,采集患儿及其父母EDTA 抗凝血各3~5 mL 进行基因分析。基因扩增: 使用BloodGen Midi Kit(CWBIO,China) 提取患儿及父母全基因组 DNA。根据人类基因组数据库中获得的ATP7A 基 因序列.应用引物设计软件Primer3,总共设计了 14 对引物,覆盖了该基因23 个编码外显子(表 1)。引物由上海英俊生物技术公司合成。根据 ATP7A 基因序列设计引物,采用PCR 方法进行扩 增。PCR 反应条件为:95℃预变性5 min;95℃变 性30 s,55℃退火30 s,72℃链延伸1 min,扩增 30 个循环;最后72℃补充延伸10 min。PCR 的体 系均为50 μL。基因序列分析:ATP7A 基因各外显 子的PCR 扩增产物,用ABI 3730XL 测序仪测序, 测序引物采用原PCR 引物。基因序列分析采用 DNASTAR 软件进行序列分析和比对。
|
|
表 1 ATP7A 基因分析引物序列 |

例1 患儿母亲再孕,于妊娠中期来院就诊, 要求进行胎儿诊断,于孕20 周时在B 超引导下 完成羊膜腔穿刺,抽取羊水20 mL,常规离心后 分取羊水细胞,使用BloodGen Midi Kit(CWBIO, China)提取胎儿羊水脱落细胞DNA,进行ATP7A 基因检测。 2 结果 2.1 临床特点
3 例患儿以顽固性惊厥为主要临床表现,分别 于2 个月、4 个月和5 个月出现抽搐,抗癫癎药物(妥 泰、德巴金、曲莱等)治疗数月无效,伴智力运 动倒退,喂养困难。3 例患儿均以原因不明“癫癎” 就诊,例3 曾因血清碱性磷酸酶升高(559.3 IU/L, 正常对照40~150 IU/L) 及25- 羟维生素D 降低 (29.2 nmol/L,正常对照47.7~144 nmol/L)被疑诊 为“佝偻病”,注射维生素D3 后2 个月未出现抽搐, 7 个半月时病情反复,频繁“屏气”样发作。来院 时3 例患儿均表现为严重智力运动障碍,不能竖 头,不认人,肤色白,双颊肥胖,头发稀黄、卷曲、 短、干燥易断,四肢肌张力低下,小头畸形,例1 前额颅缝重叠,例2 严重漏斗胸。3 例患儿的临床 特点见表 2。
|
|
表 2 3 例患儿的临床特点和主要辅助检查结果 |
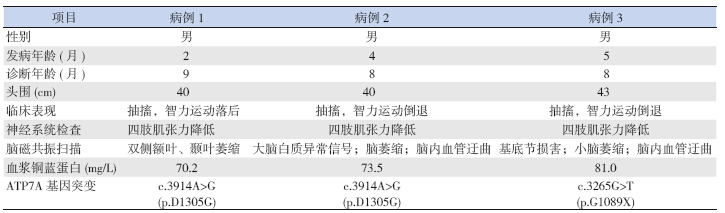
3 例患儿血液常规化验显示轻度贫血,尿常 规检查未见异常。例2 轻度肝损害,谷草转氨酶 41 IU/L(参考值0~40 IU/L),谷丙转氨酶48 IU/L (参考值0~45 IU/L)。3 例患儿血浆铜蓝蛋白均 显著降低,分别为70.2、73.5、81.0 mg/L,见表 2。 患儿父母血浆铜蓝蛋白检测均正常。 2.3 影像学检查
3 例患儿脑磁共振扫描显示不同程度的弥漫性 脑损害。例1 示双侧额叶、颞叶萎缩。例2 显示 双侧额、顶、颞区白质异常信号,大脑及小脑萎 缩,脑内血管迂曲。例3 双侧苍白球对称异常信号, 小脑萎缩,脑内血管迂曲(图 1)。
 | 图 1 患儿脑磁共振成像异常表现 例1 患儿磁共振 T2 相示双侧额叶、颞叶体积缩小,脑周间隙增宽(箭头示额叶萎 缩)。例3 患儿磁共振T2 相示双侧苍白球片状对称性长T2 信号(箭 头所示)。 |
3 例患儿均存在ATP7A 基因突变。例1 和例 2 均为c.3914A>G(p. D1305G)突变,为已报道突 变[9]。例3 为c.3265G>T(p.G1089X)突变,为未 见报道的新突变。对于c.3265G>T,我们通过千人 基因组计划(www.1000genomes.org)排除多态性, 并且应用BLAST 对比了p. G1089 在不同物种中的 保守性,发现该位点在哺乳动物中高度保守。见 表 2、图 2。
 | 图 2 ATP7A 基因测序图 例3 患儿c.3265G>T (p.G1089X)突变。箭头所指为c.3265 位点。 |
3例患儿父母ATP7A基因检测均未检出突变, 证实3 例患儿均为新生突变。 2.5 治疗与随访
3 例患儿均符合经典型Menkes 病诊断:婴 儿期起病,表现为顽固性癫癎、智力运动障碍、 特殊面容及毛发异常,血浆铜蓝蛋白明显减低, ATP7A 基因突变分析结果进一步明确了病因。由 于目前国内无合法的注射用组氨酸铜制剂,3 例患 儿均未能进行组氨酸铜注射治疗,仅以抗癫癎治 疗等对症干预为主,辅以家庭管理。
随访结果:分别进行了电话随访。例1 现在2 岁3 个月,仍不能竖头,顽固性癫癎,每5 min 抽 搐发作一次,持续数秒后自动缓解,兴奋时发作 次数增加。例2 于1 岁时发热,频繁抽搐,间断 高热10 d 后死亡。例3 现在1 岁6 个月,抽搐次 数减少,但智力运动发育停滞,消瘦。 2.6 产前诊断
例1 母亲羊水细胞染色体分析证实胎儿为 男婴,核型46,XY,ATP7A 基因测序分析未检 出c.3914A>G 突变,提示胎儿未患与例1 相同的 Menkes 病。母亲于妊娠39 周时羊膜早破,胎儿经 剖宫产出生,出生后1 min、5 min 和10 min Apgar 评分均为10 分,血清铜蓝蛋白正常,母乳喂养, 智力运动及体格发育正常,与产前诊断结果相符。 3 讨论
Menkes 病又称卷发综合征,是ATP7A 酶缺陷 导致的铜代谢障碍疾病。ATP7A 酶在人体内分布 广泛,介导铜离子的主动跨膜转运[10]。ATP7A 酶 参与结合铜离子和水解ATP,通过8 个疏水跨膜 结构域组成铜离子转运通道,将结合的铜离子转 运至细胞质[11]。ATP7A 基因突变导致ATP7A 酶缺 陷,肠黏膜对铜吸收障碍,同时细胞中的铜不能 转运至细胞间液及血液,血中铜蓝蛋白和血铜水 平降低。线粒体细胞色素C 氧化酶、超氧化岐化酶、 酪氨酸酶、赖氨酰氧化酶、多巴胺-β- 羟化酶等 铜相关性酶活性下降导致患者出现多系统功能障 碍,影响细胞呼吸、神经递质生物合成、自由基 清除、胶原合成、角蛋白合成和黑色素合成[12]。
根据患者的临床表现,Menkes 病分为经典型、 轻型和枕骨角综合征。经典型Menkes 病患者的临 床表现通常较重,患者多于3 岁内死亡。新生儿 期患者可出现黄疸、低体温、低血糖、喂养困难、 漏斗胸、腹股沟疝等。1~2 个月时开始出现特征性 的头发改变,发短、稀疏、粗糙、扭曲,面颊肥胖、 皮肤苍白。2~3 个月时可发生顽固性癫癎,随后出 现智力运动落后或倒退,骨骼异常。头颅磁共振 扫描常发现大脑及小脑萎缩、髓鞘化落后、脑白 质病、基底节损害等。动脉造影可观察到患者颅 内及颈部血管迂曲[13]。经典型患者病程呈进展性, 常在3 岁内因感染、血管并发症或神经系统功能 退化死亡。轻型患者主要表现为轻、中度的智力 运动落后,伴有毛发、皮肤改变,动脉造影可见 血管迂曲。枕骨角综合征患者头部侧位X 片见枕 骨外生骨疣,患者智力运动可正常或轻度落后, 多在成年后被发现[14]。本文3 例患儿均在婴儿期 发病,主要临床表现为顽固性癫癎和智力运动落 后、倒退,同时伴有毛发、皮肤的特征性改变, 均属于经典型Menkes 病。例1 和例2 伴有骨骼改 变(颅缝重叠和漏斗胸)。3 例患儿的脑磁共振成 像显著异常,显示脑萎缩、脑白质病和基底节损 害,其中两例患儿动脉造影显示脑血管迂曲,符 合Menkes 病脑磁共振成像表现[15]。
ATP7A 基因是目前已知的唯一与Menkes 病 相关的基因,该基因位于Xq21.1,含23 个外显 子[16]。迄今已报道约300 种不同类型的ATP7A 突 变,包括碱基置换(54%)、缺失(29%)、重 复(8%)、插入(2%)和倒位(0.1%)等。本 研究3 例患儿ATP7A 基因均检测到致病突变, 例1 和2 为c.3914A>G(p. D1305G),例3 为 c.3265G>T(p.G1089X)。2011 年Gourdon 等[9] 首 次报道了c.3914A>G(p. D1305G)。p. D1305 位 于外显子20,在不同物种中高度保守,与ATP7A 蛋白的磷酸化结构域有关,该位点突变可能导致 ATP7A 蛋白活化受限,影响铜离子跨膜转运,导 致疾病。迄今我国尚未报道过该突变,本研究为 首次报告,例1、例2 分别来自广东和河南省,无 血缘关系,父母均为非近亲结婚,推测c.3914A>G (p. D1305G)可能为中国Menkes 病患者较常见的 突变。c.3265G>T(p.G1089X)为国内外未报道的 新突变。c.3265G>T(p.G1089X)突变造成编码肽 链在1089 位提前终止,比正常肽链少了412 个氨 基酸。G1089 位于外显子16,这一区域主要负责 3例患儿父母ATP7A基因检测均未检出突变, 证实3 例患儿均为新生突变。
经典型Menkes 病病程为进展性,预后较差, 患儿多于早期儿童期死亡。该病的治疗主要依赖 铜剂补充和对症治疗、护理。由于Menkes 病患者 肠道铜吸收障碍,口服铜剂无效。国外经验证实 组氨酸铜皮下注射是控制Menkes 病的有效办法, 欧美、日本一些早期治疗的患者获得了良好的发 育[8]。目前国内尚无注射用组氨酸铜制剂,主要治 疗仍以对症治疗及家庭护理为主。本文3 例患儿 预后不良,1 例患儿在1 岁时死亡,其他两例患儿 无明显进步,例1 抽搐频率增加。
对于有Menkes 病史的家族,产前诊断是减少 疾病再发的关键措施。1994 年Tümer 等[17] 通过基 因诊断,首次成功地进行了Menkes 病产前诊断。 Menkes 病产前诊断的方法主要有以下三种:基因 分析、检测羊水细胞对64Cu 的摄取量以及检测绒 毛细胞的铜离子浓度[18-20]。由于检测方法的限制 以及生化检测的不稳定性,基因分析技术更为普 遍、可靠。在先证者基因突变明确的情况下,对 羊水细胞进行ATP7A 基因分析可明确胎儿是否为 Menkes 病患者。本研究中例1 母亲之胎儿为男性, 羊水细胞ATP7A 基因分析未见异常,胎儿出生后 发育正常,验证了产前诊断结果。
本研究总结和分析了3 例经典型Menkes 病患 儿的临床表现和实验室诊断,发现了一种ATP7A 基因新突变c.3265G>T(p.G1089X),首次从中国 患者检出了c.3914A>G(p. D1305G),进一步丰 富了我国Menkes 病的基因突变谱,并帮助一个家 系进行了胎儿的产前诊断,为我国大陆地区首例 Menkes 病产前诊断经验。
| [1] | Menkes JH, Alter M, Steigleder GK, et al. A sex-linked recessive disorder with retardation of growth, peculiar hair and focal cerebral and cerebellar degeneration[J].Pediatrics, 1962, 29 (5): 764-779. |
| [2] | Gu YH, Kodama H, Shiga K, et al. A survey of Japanese patients with Menkes disease from 1990 to 2003: incidence and early signs before typical symptomatic onset, pointing the way to earlier diagnosis[J].J Inherit Metab Dis, 2005, 28 (4): 473-478. |
| [3] | 王晓慧, 吕俊兰, 张礼萍, 等. Menkes 病三例临床及实验室 特点[J].中华儿科杂志, 2009, 47 (8): 604-606. |
| [4] | 赵程峰, 王静敏, 王菊莉, 等. Menkes 病患儿ATP7A 基因突 变及X 染色体失活分析[J].中国伤残医学, 2013, 21 (5): 37-41. |
| [5] | 麻宏伟, 陈丽英, 张海娟, 等. Menkes 病一家系二例[J].中 华医学遗传学杂志, 2001, 18 (4): 258. |
| [6] | Kaler SG. Diagnosis and therapy of Menkes syndrome, a genetic form of copper deficiency[J].Am J Clin Nutr, 1998, 67 (5): 1029- 1034. |
| [7] | Tümer Z. An Overview and update of ATP7A mutations leading to menkes disease and occipital horn syndrome[J].Hum Mutat, 2013, 34 (3): 417-429. |
| [8] | Christodoulou J, Danks DM, Sarkar B, et al. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients[J].Am J Med Genet, 1998, 76 (2): 154-164. |
| [9] | Gourdon P, Liu XY, Skjorringe T, et al. Crystal structure of a copper-transporting PIB-type ATPase[J].Nature, 2011, 475 (7354): 59-64. |
| [10] | Lutsenko S, LeShane ES, Shinde U. Biochemical basis of regulation of human coppertransporting ATPases[J].Arch Biochem Biophys, 2007, 463(2): 134-148. |
| [11] | Palmgren MG, Nissen P. P-type ATPases[J].Annu Rev Biophys, 2011, 40: 243-266. |
| [12] | Horn N, Tümer Z. Menkes disease and the occipital horn syndrome[M]// Royce PM, Steinmann B. Connecitive Tissue and its Heritable Disorders: Molecular, Genetic, and Medical Aspects. 2nd ed. New York: John Wiley and Sons Inc, 2002: 651-685. |
| [13] | 黄琼辉, 王静敏, 吴晔, 等. Menkes 病临床及ATP7A 基因 突变和拷贝数改变分析[J].实用儿科临床杂志, 2012, 27 (8): 570-573. |
| [14] | 邓艳华, 王静敏, 牛争平, 等. Menkes 病患儿ATP7A 基因突 变分析[J].实用儿科临床杂志, 2007, 22 (24): 1877-1879. |
| [15] | 程晓悦, 肖江喜, 袁新宇, 等. Menkes 病的MR 影像表现[J].中华放射学杂志, 2013, 47 (7): 599-602. |
| [16] | Tümer Z, Vural B, Tunnesen T, et al. Characterization of the exon structure of the Menkes disease gene using vectorette PCR[J].Genomics, 1995, 26 (3): 437-442. |
| [17] | Tümer Z, Tunnesen T, Bohmann J, et al. First trimester prenatal diagnosis of Menkes disease by DNA analysis[J].J Med Genet, 1994, 31 (8): 615-617. |
| [18] | Horn N. Menkes' X-linked disease: prenatal diagnosis and carrier detection[J].J Inherit Metab Dis, 1983, 6(1): 59-62. |
| [19] | Heydorn K, Damsgaard E, Horn N. Accumulated experience with prenatal diagnosis of Menkes disease by neutron activation analysis of chorionic villi specimens[J].Biol Trace Elem Res, 1999, 71-72; 551-561. |
| [20] | Das S, Whitney S, Taylor J, et al. Prenatal diagnosis of Menkes disease by mutation analysis[J].J Inher Metab Dis, 1995, 18 (3): 364-365. |