 2014, Vol. 16
2014, Vol. 16



2. 广西卫生职业技术学院, 广西 南宁 530002
选取2009 年1 月至2012 年7 月因发育迟缓、 智力低下、生殖器外观异常、多发畸形等临床指 征就诊于我院的患儿601 例为研究对象,其中男 352 例,女249 例,年龄15 h 至16 岁,平均年龄 7 岁。 1.2 研究方法
用肝素抗凝管采集患儿外周血3 mL,接种全 血15~20 滴于培养基中,培养基为湖南湘雅基因 有限公司或广州拜迪生物医药有限公司产品。于 37±5℃条件下培养66~72 h 后进行常规染色体制 备及G 显带。每例分析5 个核型,计数25 个中期 分裂相,嵌合体加倍计数分析。染色体分析仪器 设备为MetaSystems 染色体自动扫描分析系统(德 国ZEISS)。根据人类遗传学国际命名体制(ISCN 2009)对染色体核型命名。 2 结果 2.1 异常核型分析
在601 例受检者中,共检出329 例异常核型, 检出率为54.7%,其中数目异常317 例(96.4%), 结构异常12 例(3.6%)。12 例结构异常核型中, 共检出8 种人类染色体新核型,涉及13 条常染色 体,其中衍生染色体3 例,易位4 例,倒位1 例(表 1)。
| 表 1 329 例染色体异常核型分析 |
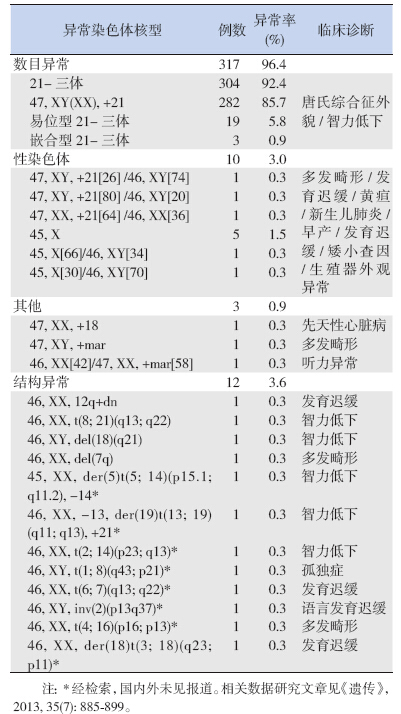
为了进一步阐述染色体异常是导致发育迟缓、 智力低下、生殖器外观异常、多发畸形等疾病的 重要原因,以下从病例中选出典型的2 例新核型 进行分析。
病例1:患儿,女,5 岁,智力低下。染色体 核型为45,XX,der(5)t(5; 14)(p15.1; q11.2),-14(图 1), 其父母染色体未查。
 |
图 1 病例1 染色体核型 患儿,女,5 岁,智力低下, 染色体核型为45,XX,der(5)t(5;14)(p15.1; q11.2),-14,箭头所示为衍 生染色体。 |
病例2: 患儿,男,5 岁,智力低下,头 颅CT 示枕大池囊肿,小脑发育不全,侧脑室增宽。外院核型分析结果显示染色体核型为47,XY, +der(14)t(2; 14)(p23; q11)。母亲,27 岁,染色体核 型为46,XX,t(2; 14)(p23; q13),见图 2。
 |
图 2 病例2 母亲的染色体核型 病例2 母亲的染色体 核型为46,XX,t(2; 14)(p23; q13),箭头所示为平衡易位点。 |
根据送检单提供的临床诊断信息统计,因疑 似唐氏综合征查因276 例,确诊染色体异常248 例; 因发育迟缓或矮小查因68 例,确诊染色体异常36 例;因智力低下送检54 例,确诊染色体异常10 例; 因生殖器外观异常送检42 例,确诊染色体异常2 例,因新生儿肺炎送检38 例,确诊染色体异常16 例;此外,早产或黄疸确诊染色体异常7 例,因 听力异常、先天性心脏病、语言发育迟缓、孤独 症确诊染色体异常各1 例,见表 2。
| 表 2 601 例儿童患者临床诊断及异常核型分布 [n(%)] |
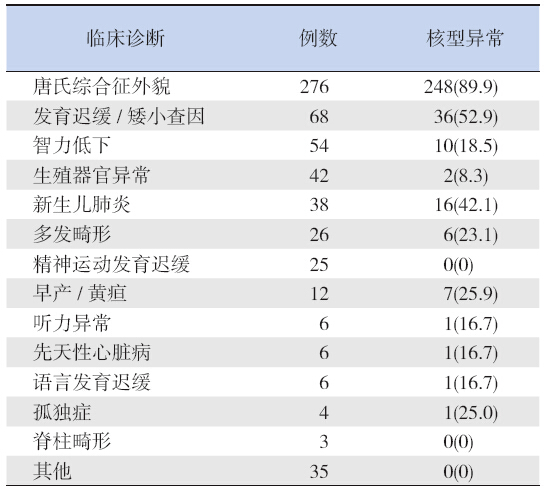
在儿童遗传咨询中,21- 三体是最常见的染色 体异常类型。本研究中21- 三体综合征患儿例数 占异常核型总数的92.4%(304/329)。唐氏综合征属于遗传咨询中较为容易识别的染色体病,患 者具有特殊面容,如圆脸、眼距宽、睑裂上斜、 内眦赘皮、塌鼻梁、耳位低、通贯掌等。常伴有 智力低下、发育迟缓、先天性心脏病,胃肠道畸 形等。本研究显示,临床诊断为唐氏综合征外貌 的276 例患者中,89.9%(248/276)确诊为21- 三 体。唐氏综合征决定区域(down syndrome critical region,DSCR)位于21q22,片段大小约5.4 Mb, 这个区域的重复可导致个体出现典型的唐氏综合 征的临床表型[3]。Ronan 等[4] 报道一个唐氏综合 征患者家系,采用染色体核型分析及中期荧光原 位杂交技术检测3 位患者未见异常,但最终采用 间期荧光原位杂交和比较基因组杂交技术检出在 21q22.13-q22.2 区存在有4.3 Mb 的微重复。因此儿 童遗传咨询的病例中,医生提供的临床信息可为 实验室工作人员研究的方向提供很大帮助。
先天畸形、智力低下、发育迟缓、语言障碍 等症状也是儿童咨询病例常见的主诉病因。病例 1 先证者,5 岁,主诉为智力低下,核型分析为 45,XX,der(5)t(5; 14)(p15.1; q11.2),-14,该衍生染色 体来源于5p15.1 至5per 片段丢失和14 号染色体 部分随体丢失后重排。5 号染色体短臂的部分缺失 可导致“猫叫综合征”,临床上猫叫样哭声和小 头畸形可随年龄增长变得不典型。由于其双亲未 做染色体核型分析,推测亲代中可能有一方为t(5; 14 ) 的非同源染色体平衡易位者。在第一次减数分 裂的中期形成四射体,分离与交换后,理论上可 形成18 种配子。不平衡易位胎儿有2%~10% 的可 能性幸存出生,均伴有多发畸形、智力低下、发 育迟缓等原因[5]。病例2 母亲核型分析为46,XX, t(2; 14)(p23; q13),患儿为47,XY,+der(14)t(2; 14)(p23; q11)。由此可以推测先证者的重组染色体多出来 的片段是母源性的衍生染色体。鉴于该类型患者 细胞遗传学发生机制复杂,往往需要溯源检查其 亲代的染色体,为患儿治疗的指导及父母再次妊 娠提供帮助。表1 中的46,XX,-13,der(19)t(13; 19) (q11; q13),+21 及46,XX,der(18)t(3; 18)(q23; p11) 为 类似的情形,应进一步检查父母双方的核型。表1 中的另外几例非衍生染色体的异常核型:46,XY, t(1; 8)(q43; p21)、46,XX,t(6; 7)(q13; q22)、46,XY, inv(2)(p13q37)、46,XX,t(4; 16)(p16; p13),幼儿临床 表现为孤独症、发育迟缓、语言发育迟缓、多发畸形等。这4 例患儿父母核型均为正常,推测是 新发突变所致。
智力低下患儿发病机制主要是由于受到多种 因素的影响导致大脑发育和成熟受到损伤所致, 如孕期或出生时感染、营养不良、有毒物质侵害等, 患儿临床表现为脑瘫、孤独症、认知和适应能力 受损等,在人群中的发病率为1%~3%[1]。采用不 同的技术检测显示,17%~41% 的原因是由于遗传 学因素导致的,如染色体异常、单基因病、基因 的其他异常等[6]。往往精神发育迟缓或脑瘫的病例, 不易检出染色体异常。本研究中,29 例精神发育 迟缓或脑瘫的病例均未见染色体异常。临床医生 一般会对该类型患儿进行认知及行为能力评估、 家系调查等。排除脆性X 综合征后,若进一步的 采用荧光原位杂交、MLPA、比较基因组杂交诊断 技术,可诊断出5%~20% 的患者存在微小异常[7]。 有研究显示,1q21.1、15q13.3 及16p11.2 为精神 发育迟缓、孤独症、癫癎、精神分裂症等疾病的 的染色体重组热区[8]。临床医生尽可能提供的详尽 信息,如22q11 微缺失综合征常见的临床表现为 心脏畸形、腭裂及低血钙症等,眼距、耳朵的形状、 皮肤的性状、头围、四肢、生长参数等,都将便 于实验室人员甄别患者是属于哪种已知的微缺失 或微重复的综合征,而更为先进的基因技术应用 将为遗传咨询和产前诊断提供更有力的诊断手段。
本实验室检出性染色体异常的类型主要为 Turner 综合征及其嵌合体。Turner 综合征发生率为 1 : 2 000,异常表型体现为身材矮小、性腺发育不 全、闭经、颈蹼等[9]。主要是由于父源性的减数分 裂中精子发生染色体丢失所致。嵌合型的Turner 综合征,即45,X 和另一个细胞系的嵌合体。患 者临床表型呈多样化,可为两性畸形、外生殖器 模糊,表型未见异常等。本研究中有两例患者分 别因先天性尿道下裂和矮小查因行染色体检查, 结果为45,X/46,XY。在生殖器官异常的病例中, 染色体异常检出率为8.33%(2/42),未见异常 的病例可进一步检测Y 染色体性别决定区(sex determining region of Y chromosome,SRY)。SRY 位 于Yp11.32,减数分裂期,Y 染色体可与X 染色体 发生配对及重组,20% 性分化与染色体核型不符 的患者是由于SRY 在重组中丢失或获得所致的[10]。 因此对疑似性发育异常的病例,不论是获得功能 (46,XX 性逆转和真两性畸形)还是丧失功能病 例(46,XY 性发育不全和真两性畸形),SRY 的 检测可以进一步的明确病因[11]。
综上所述,染色体异常是导致儿童发育迟缓、 智力低下、生殖器官异常、多发畸形等疾病的重 要原因。患有染色体病的患儿,往往生活不能自理, 生命质量不高,给社会及家庭带来沉重的负担。 借助遗传学诊断技术对疑似患有遗传病的患儿查 找出病因,对患儿的治疗及其父母的再次妊娠具 有积极的指导意义。
志谢:本文8 种人类染色体核型经中南大学医学 遗传学实验室夏家辉院士、邬玲仟教授、戴和平教授、 龙志高教授等鉴定,为人类染色体新核型。特此志谢!
| [1] | Stevenson RE, Holden KR, Rogers RC, et al. Seizures and X-linked intellectual disability[J]. Eur J Med Genet, 2012, 55(5): 307-312. |
| [2] | Mefford HC, Batshaw ML, Hoffman EP, et al. Genomics, intellectual disability, and autism[J]. N Engl J Med, 2012, 366(8): 733-743. |
| [3] | Lana-Elola E, Watson-Scales SD, Fisher EM, et al. Down syndrome: searching for the genetic culprits[J]. Dis Model Mech, 2011, 4(5): 586-595. |
| [4] | Ronan A, Fagan K, Christie L, et al. Familial 4.3 Mb duplication of 21q22 sheds new light on the Down syndrome critical region[J]. J Med Genet, 2007, 44(7): 448-451. |
| [5] | Keymolen K, Staessen C, Verpoest W, et al. Preimplantation genetic diagnosis in female and male carriers of reciprocal translocations: clinical outcome until delivery of 312 cycles[J]. Eur J Hum Genet, 2012, 20(4): 376-380. |
| [6] | Bernardini L, Alesi V, Loddo S, et al. High-resolution SNP arrays in mental retardation diagnostics: how much do we gain?[J]. Eur J Hum Genet, 2010, 18(2): 178-185. |
| [7] | Tucker T, Montpetit A, Chai D, et al. Comparison of genomewide array genomic hybridization platforms for the detection of copy number variants in idiopathic mental retardation[J]. BMC Med Genomics, 2011, 25(4): 25. |
| [8] | Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders and common disease[J]. Curr Opin Genet Dev, 2009, 19(3): 196-204. |
| [9] | 王红, 金煜炜, 翟宇晋, 等. Turner 综合征232 例染色体核 型、诊断年龄和身高分析[J]. 中华儿科实用临床杂志, 2013, 28(8): 596-599. |
| [10] | Larson A, Nokoff NJ, Travers S. Disorders of sex development: clinically relevant genes involved in gonadal differentiation[J]. Discov Med, 2012, 14(78): 301-309. |
| [11] | 向萍霞, 戴翔, 冷培, 等. SRY 基因检测在儿童性发育疾病 诊断中的应用[J]. 中国当代儿科杂志, 2013, 15(7): 555-558. |