 2014, Vol. 16
2014, Vol. 16



2. 广东省妇幼保健院检验科, 广东 广州 510010
解偶联蛋白2(uncoupling protein 2,UCP2)是 存在于线粒体内膜的质子转运体,广泛表达于各 种器官组织中,包括脾、肺、肠、脑、心肌、胰 岛细胞和白色脂肪组织[1]。UCP2 在限制线粒体活 性氧(reactive oxygen species,ROS)产生和能量生 成调控方面具有关键作用。UCP2 可参与各种病理 生理过程,包括胰岛素的分泌、肥胖、糖尿病或 神经细胞的活动及缺血再灌注损伤等[2]。最新研究 发现UCP2 可以通过介导ROS 的产生参与细胞内 炎症反应的调控[3]。炎症损伤是脓毒症心肌损伤 的重要病理生理机制,目前对于UCP2 在脓毒症心 肌病的具体作用机制国内还尚未有报道,为了探 索UCP2 参与脓毒症心肌病的可能发病机制,本研 究拟运用脓毒症大鼠血清诱导刺激大鼠心肌细胞 株H9C2 细胞建立脓毒症心肌损伤体外模型,通过 RNA 干扰技术影响UCP2 基因的表达,探讨UCP2 对脓毒症心肌损伤MAPK、NF-κB 炎症通路以及炎 症因子TNF-α、IL-1β 的影响。 1 材料与方法 1.1 动物与材料
健康成年清洁级(SPF)雄性Sprague-Dawley 大鼠30 只(广东省医学实验动物中心提供), 体重250~300 g; 大肠埃希菌脂多糖(LPS, Escherichiacoli0111:134,美国Sigma 公司); UCP2 多克隆羊抗体,β-actin 抗体(Santa Cruz Biotechnology,Inc );磷酸化p38MAPK(p-p38MAPK) 和核转录因子(NF-κB)抗体(购于Cell Signaling Technology,Inc.);Trizol,PrimeScript™ RT reagent Kit,SYBR® Premix Ex Taq™( 大连宝生生物公 司);ABI 7500 荧光定量PCR 仪、ABI9700PCR 仪(美国应用生物公司);阳离子脂质体hilymax (同仁化学公司)。根据GenBank 的UCP2 基因 序列(NM_019354.2),由广州锐博公司设计并 合成 3 对siRNA 并设计阴性对照siRNA。UCP2- siRNA1 上游GCACUGUCGAAGCCUACAAdTdT, 下游dTdTCGUGACAGCUUCGGAUGUU;UCP2- siRNA2 上游CCUCAUGACAGACGACCUCdTdT, 下游dTdT GGAGUACUGUCUGCUGGAG;UCP2- siRNA3 上游CCUGGAACGUAGUAAUGUUdTdT, 下游d T d T G G A C C U U G C A U C A U U A C A A 。 UCP2、β-actin、TNF-α、IL-1β 引物由上 海英伟捷基公司设计并合成。UCP2 引物序 列: 上游ACCATTGCACGAGAGGAAGG,下 游TCTTGACCACATCAACGGGG;β-actin 引 物序列: 上游GCAGGAGTACGATGAGTCCG, 下游ACGCAGCTCAGTAACAGT;TNF-α 引物 序列: 上游AACACACGAGACGCTGAAGT,下 游TCCAGTGAGTTCCGAAAGCC;IL-1β 引物序 列: 上游CCTTGTCGAGAATGGGCAGT,下游 TTCTGTCGACAATGCTGCCT。 1.2 脓毒症大鼠和正常大鼠血清制备
实验前大鼠禁食12 h,自由饮水。脓毒症大 鼠血清制备参照文献方法[4],内素素 (大肠埃希 菌脂多糖,LPS)10 mg/kg 腹腔注射,6 h 后无菌 心脏穿刺采血,4℃静置30 min,800 r/min 离心后 生物安全柜内收集上清液。-80℃保存待用。正常 大鼠血清取自不经任何干预大鼠,方法同上。 1.3 细胞培养转染
H9C2 细胞由南方医科大学珠江医院心内科保 存,转染前1 d 将大约4×105 个细胞接种于6 孔 板中,当细胞覆盖达60%~80%时采用阳离子脂 质体分别转染siRNA。转染方法参照hilymax 说明 书,转染后37℃培养箱培养,4~6 h 后换含血清培 养液继续培养,先从由锐博公司合成的siRNA1、 siRNA2、siRNA3 序列筛选出有效干扰序列,转染 后进行后续实验。 1.4 细胞分组及处理
体外培养大鼠心肌细胞株(H9C2)随机分为 5 组:空白对照组、正常血清组、10%脓毒症血清 刺激12 h 组(脓毒症血清组)、UCP2-siRNA 干扰 +10%脓毒症血清刺激12 h 组(UCP2-siRNA+ 脓 毒症血清组)、阴性siRNA 转染+10%脓毒症血 清刺激12 h 组(阴性siRNA + 脓毒症血清组), 每组设3 个复孔。(1)空白对照组不经处理,大 鼠H9C2 心肌细胞株在含10% 胎牛血清DEME- 高 糖培养基下培养箱培养;(2)正常血清处理组: 待当细胞生长至70%~80% 密度时,加入10% 正 常大鼠血清培养12 h;(3)10%脓毒症血清刺激 12 h 组:待当细胞生长至70%~80% 密度时加入 10% 脓毒症大鼠血清培养12 h;(4)阴性siRNA 转染+10%脓毒症血清刺激12 h 组:待细胞生长 至70%~80% 密度时先行阴性si-RNA 转染,转染24 h 后加入10% 脓毒症大鼠血清培养12 h; (5)UCP2-siRNA 干扰+10%脓毒症血清刺激12 h 组:待细胞生长至70%~80% 密度时先行UCP2si- RNA 转染,转染24 h 后加入10% 脓毒症大鼠血清 培养12 h。 1.5 RT-PCR 检测UCP2、TNF-α、IL-1βmRNA 的表达
采用TRizol 试剂提取H9C2 心肌细胞总 RNA,ND-100 分光光度计测得RNA 的纯度和含 量。取500 ng RNA 进行逆转录反应合成cDNA。 然后以20 μL 反应体系SyberGreen 荧光定量PCR 检测。PCR 热循环参数: 95℃预变性30 s,95℃ 变性5 s,60℃退火34 s,40 个循环。用ΔΔCt 法 计算mRNA 的变化。 1.6 Western blot 检测UCP2、p-p38MAPK、NF-κB 蛋白表达
采用全蛋白提取试剂盒提取H9C2 心肌细胞 总蛋白,BCA 法测蛋白浓度。用凝胶加样缓冲液 将各管蛋白样品煮沸5 min。按每孔20 μg 蛋白量 加样SDS-PAGE 电泳,电泳结束后将凝胶取出, 根据Marker 切胶、转膜,丽春红预染。5%脱脂 奶粉的TBST 封闭室温2 h,TBST 漂洗3 次,每 次10 min,羊抗UCP2(1 : 500)、兔抗beta-actin (1 : 1 000)、兔抗磷酸化p38MAPK(1 : 1 000)和 核转录因子NF-κB(1 : 1 000)抗体孵育,4℃摇床 过夜,TBST 洗3 次,每次10 min,(1 : 7 500)二 抗室温孵育1 h,TBST 洗3 次,每次10 min 后使 用ECL 发光法暗室显影,扫描分析。以β-actin 作为内参照,分析UCP2、p-p38MAPK、NF-κB 条 带灰度变化。 1.7 统计学分析
应用SPSS 18.0 软件对数据进行分析,数据 以均数± 标准差(x±s)表示,多组样本之间 比较采取方差分析,方差不齐时用Welch 校正检 验。多个样本均数间的多重比较采用LSD 法或 Dunnetts T3 法检验,P<0.05 为差异有统计学意义。 2 结果 2.1 Western blot 和RT-PCR 筛选最有效的siRNA
siRNA1、siRNA2、siRNA3 等3 对干扰序列进 行干扰,3 对序列均可以明显沉默UCP2 mRNA 表 达(P<0.01),其中siRNA2 序列对UCP2 mRNA 抑制效果最强,即siRNA2 干扰H9C2 心肌细胞后, 干扰效果最强。荧光定量PCR 结果显示siRNA 2 组序列UCP2 相对表达为(0.286±0.010),明显 低于对照组(1.038±0.044),差异有统计学意义 (F=97.86,P<0.01)。同时蛋白水平显示siRNA 2 序列最有效,故后续实验均以siRNA 2 为靶序列 进行。见图 1,2。
 |
图 1 荧光定量PCR 检测各组细胞UCP2 mRNA 沉 默效率(n=3) 注:a 示与对照组相比,P<0.01;b 示与阴性 siRNA 组比较,P<0.01;c 示与siRNA1 组比较,P<0.01;d 示与 siRNA2 组比较,P<0.01。 |
 |
图 2 UCP2 siRNA2 序列干扰效果Western blot 验证图 |
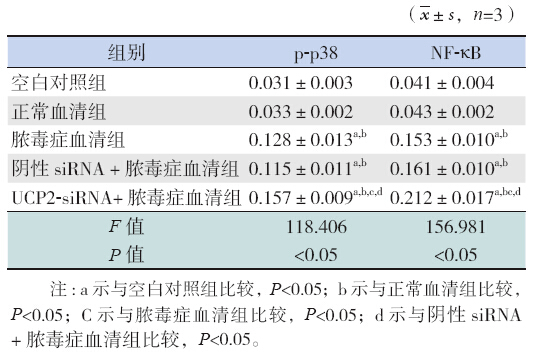
脓毒症血清刺激H9C2 心肌细胞株后细胞 p-p38、NF-κB 蛋白表达与对照组和正常血清组 相比均明显增加(P<0.05),其中UCP2-siRNA+ 脓毒症血清组表达较脓毒症大鼠血清组更加显著 (P<0.05),见表 1,图 3。
| 表 1 各组细胞p-p38 和NF-κB 蛋白表达的变化 |
 |
图 3 各组细胞p-p38 和NF-κB 蛋白表达 1:空白 对照组;2:正常大鼠血清组;3:脓毒症血清组;4:阴性siRNA 转染+ 脓毒症血清组;5:UCP2-siRNA 干扰+ 脓毒症血清组。 |
荧光定量PCR 检测显示,脓毒症大鼠血清刺 激后各组细胞TNF-α、IL-1βmRNA 表达均明显上 调。与正常对照、正常血清组相比,脓毒症血清 组、阴性siRNA+ 脓毒症血清组、UCP2-siRNA+ 脓 毒症血清组TNF-α mRNA 相对表达(2.74±0.29, 2.81±0.39,4.72±0.27)均明显增高,差异均有统 计学意义(P<0.01),其中UCP2-siRNA+ 脓毒症血 清组与脓毒症血清组和阴性siRNA+ 脓毒症血清组 比较有明显增高,差异统计学意义(P<0.01)。脓 毒症血清组、阴性siRNA+ 脓毒症血清组、UCP2- siRNA+ 脓毒症血清组IL-1βmRNA 相对表达分别 为(11.8±1.6,12.8±2.6,13.2±0.5) 均明显高 于正常对照组和正常血清组(P<0.01),脓毒症血 清组、阴性siRNA+ 脓毒症血清组与UCP2-siRNA+ 脓毒症血清组之间相比差异均无统计学意义。见 图 4。
 |
图 4 荧光定量PCR 各组TNF-a、IL-1βmRNA 的 表达(x±s,n=3) A:正常对照组;B:正常大鼠血清组;C: 脓毒症血清组;D:阴性siRNA + 脓毒症血清组;E:UCP2-siRNA + 脓毒症血清组。注:a 示与对照组相比,P<0.01,b 示与正常血 清组比较,P<0.01。c 示与脓毒症血清组比较,P<0.01;d 示与阴 性siRNA + 脓毒症血清组比较,P<0.01。 |
脓毒症心肌病是脓毒症的严重并发症之一, 其主要表现包括有可逆性的双心室扩张、心肌射 血分数下降,对液体复苏和正性肌力药物的反应 下降,同时出现血液中肌钙蛋白、BNP 酶学等心 肌标记物上升[5]。目前关于脓毒症心肌病的病理生 理机制仍不明确,有研究认为一氧化氮、线粒体 功能障碍、循环补体和细胞因子在其中发挥关键 作用[6]。当细菌所致脓毒症时,G- 菌的LPS 或G+ 菌的肽聚糖和磷壁酸通过与模式识别受体(PRRs) 识别,进而激活单核- 巨噬细胞系统NF-κB 和丝 裂原激活的蛋白激酶MAPK 等炎症通路,随之大 量炎症介质和细胞因子释放(如IL-6、TNF-α、 IL-1 等)[7-9]。血液循环中的内毒素、炎症介质和 细胞因子,一方面可以直接对心肌细胞造成损害, 另外可以激活心肌细胞内MAPK,NF-KB 等炎症 通路,影响心肌功能[10]。既往研究结果显示, p38MAPK,NF-KB 信号转导通路在脓毒症心肌组 织中激活,并促发TNF-α 和IL-1β 等炎症介质的 释放,阻断两者信号转导通路可改善心肌损伤[11]。 本研究也发现,在脓毒症大鼠血清刺激作用下, H9C2 心肌细胞株内p-p38MAPK 和NF-κB 表达发 生上调; TNF-α mRNA、IL-1βmRNA 也发生上调。
研究证实UCP2 可以调控细胞内炎症反应, UCP2 基因敲除小鼠在LPS 作用下其血清TNF 和 干扰素α 的水平较野生型小鼠更高,同时巨噬细 胞的NF-κB 表达也更高,这可能与UCP2 通过影 响细胞内ROS 的产生起作用的[12]。最近Chul 等[13] 研究发现,能够有效下调细胞内炎症反应的一个 细胞内信号分子细胞核孤儿受体—微小异源二聚 体伴侣基因(small heterodimer partner,SHP) 是 通过UCP2 影响细胞内氧化应激信号起作用的。 UCP2 基因对于脓毒症心肌病作用的研究还未见 报道。在脓毒症血清诱导刺激下大鼠心肌细胞株 细胞内炎症通路被激活,并且炎症因子TNF-α、 IL-1β 在mRNA 水平上表达上调。进一步研究发 现RNA 干扰技术沉默H9C2 细胞UCP2 基因后, 细胞内p38MAPK 活性和NF-κB 激活加强,TNF-α 转录增强。提示UCP2siRNA 转染可以促进心肌细 胞内炎症反应,因此推测,UCP2 在脓毒症心肌组 织表达可能能够反馈下调心肌细胞内炎症通路和 氧化应激损伤。但是本研究未能验证UCP2 如何影 响炎症通路的形成。在细胞模型方面,本研究只 选用单一的基因干扰,严格来说需同时使用过表 达技术双方面验证甚至需要使用基因敲除或转基 因动物进一步验证。所以就UCP2 在脓毒症心肌病 的具体机制还有待进一步研究。
| [1] | Donadelli M, Dando I, Fiorini C, et al. UCP2, a mitochondrial protein regulated at multiple levels[J]. Cell Mol Life Sci, 2014, 71(7): 1171-1190. |
| [2] | Azzu V, Jastroch M, Divakaruni AS, et al. The regulation and turnover of mitochondrial uncoupling proteins[J]. Biochim Biophys Acta, 2010, 1797(6-7): 785-791. |
| [3] | Emre Y, Nübel T. Uncoupling protein UCP2: When mitochondrial activity meets immunity[J]. FEBS Letters, 2010, 584(8): 1437-1442. |
| [4] | Fink MP, Heard SO. Laboratory models of sepsis and septic shock[J]. J Surg Res, 1990, 49(2): 186-196. |
| [5] | Vieillard-Baron A. Septic cardiomyopathy[J]. Annals of Intensive Care, 2011, 1(1): 6. |
| [6] | Flynn A, Chokkalingam Mani B, Mather PJ. Sepsis-induced cardiomyopathy: a review of pathophysiologic mechanisms[J]. Heart Fail Rev, 2010, 5(6): 605-611. |
| [7] | Shimaoka M, Park EJ. Advances in understanding sepsis[J]. Eur J Anaesthesiol Suppl, 2008, 42(25): 146-153. |
| [8] | Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis[J]. Nat Rev Immunol, 2008, 8(10): 776- 787. |
| [9] | 黄卫东, 郭景涛, 刘喜, 等. 全身炎症反应综合征患儿中性 粒细胞淋巴细胞CD11b 表达意义[J]. 中国当代儿科杂志, 2009, 11(7): 541-542. |
| [10] | Zhang T, Feng Q. Nitric oxide and calcium signaling regulate myocardial tumor necrosis factor-a expression and cardiac function in sepsis[J]. Can J Physiol Pharmacol, 2010, 88(2): 92- 104. |
| [11] | Yang M, Wu J, Martin CM, et al. Important role of p38 MAP kinaseNF-kB signaling pathway in the sepsis-induced conversion of cardiac myocytes to a proinflammatory phenotype[J]. Am J Physiol Heart Circ Physiol, 2008, 294(2): H994-H1001. |
| [12] | Yushi B, Hiroki O, Xu B, et al. Persistent Nuclear Factor-kB Activation in Ucp2-- Mice Leads to Enhanced Nitric Oxide and Inflammatory Cytokine Production[J]. J Biol Chem, 2005, 13, 280(19): 19062-19069. |
| [13] | Chul SY, Jae MY, Jwa JK, et al. Small heterodimer partnertargeting therapy inhibits systemic inflammatory responses through mitochondrial uncoupling protein 2[J]. PLoS ONE, 2013, 8(5): e63435. |