 2015, Vol. 17
2015, Vol. 17

2. 中国医学科学院北京协和医学院血液病医院血液学研究所 血液病理诊断中心, 天津 300020


2008 年WHO 造血与淋巴组织肿瘤分类标 准[1]将儿童骨髓增生异常综合征(myelodysplastic syndrome,MDS)分为难治性血细胞减少(refractory cytopenia of childhood,RCC)、难治性贫血伴原 始细胞增高(refractory anemia with excess blasts, RAEB)、难治性血细胞减少伴原始细胞增高-转化型(refractory anemia with excess blasts in transformation,RAEB-t)。
RCC 是一个新提出的暂定分类,其诊断标准 为:持续的血细胞减少伴外周血原始细胞 <2%, 骨髓原始细胞 <5%,且一系病态造血比例大于 10% 或两系病态造血,并除外其他骨髓衰竭性疾 病及继发性 MDS [1]。这一亚型的提出是基于原始 细胞不高的儿童 MDS 具有一些与成人 MDS 不同的 临床特征,包括:成人 MDS 分类中的难治性贫血 伴环状铁粒幼细胞贫血,MDS 伴单纯 5q-在儿童 罕见[2];以单纯贫血(即成人的难治性贫血)起病 在儿童少见,多伴有中性粒细胞和(或)血小板 减少[3, 4];骨髓增生减低更常见。因此,成人“低危” MDS 的分类不适用于儿童 MDS 患者。
RCC 包含了骨髓增生重度减低伴轻度病态造 血的患儿和骨髓增生程度正常伴明显病态造血的 符合成人难治性血细胞减少伴多系病态造血的患 儿。骨髓增生减低的 RCC 与另一大类骨髓衰竭 性疾病—获得性再生障碍性贫血(aplastic anemia, AA),特别是与非重型 AA 的鉴别诊断存在困难: 两者均表现为骨髓增生减低,病态造血不易识别, 无特定的免疫学和细胞遗传学标志。RCC 与非重 型 AA 均为慢性病程,需要长期随访才能判断两者 临床转归。从发病机制上讲,RCC 是一种恶性克 隆性疾病,有着潜在的向白血病转化的风险,而 非重型 AA 是一种主要由免疫介导的良性疾病,分 析两者的临床特征及转归,对临床治疗选择具有 重要的意义。本研究回顾性分析儿童 RCC 的临床 特征、疗效及临床转归,现报告如下。 1 资料与方法 1.1 研究对象
研究对象为1990年1月至2013年6月我院 门诊及住院符合以下标准的患者:(1)年龄 0~14 岁之间;(2)初始诊断为非重型 AA;(3)初诊 时的骨髓细胞形态学及骨髓病理学资料均完整; (4)至我院前未进行治疗。
排除标准:(1)初始诊断为重型 AA/ 超重型 AA 的患者;(2)具有可疑的先天性骨髓衰竭的 体征或实验室检查结果,如肢体畸形和(或)丝 裂霉素断裂实验阳性和(或)单个细胞凝胶电泳 实验阳性;(3)有可疑的血液系统疾病和(或) 肿瘤家族史;(4)既往有化疗药物治疗史和(或) 放射线接触史;(5)怀疑继发于感染、维生素缺 乏或自身免疫病的血细胞减少。
非重型 AA 诊断标准参照文献[5, 6];重型及超 重型 AA 诊断标准参照文献[6]。
该研究经中国医学科学院北京协和医学院血 液病医院血液学研究所伦理委员会批准。 1.2 骨髓形态学回顾性分析
患儿骨髓细胞形态学和骨髓组织病理学由 3 名专家重新评估。诊断流程及标准参考既往研 究[7, 8, 9, 10]。骨髓细胞形态学、骨髓病理学样本根据 FAB(French-American-British Cooperative Leukemia Working Group)和 EWOG-MDS(the morphology group of the European Working Group MDS in children)标准 评估[11, 12]。RCC 的诊断根据 WHO 2008 标准[1]。骨 髓纤维化分级根据既往报道[13]。 1.3 染色体核型分析
骨髓细胞染色体核型分析采用R显带技 术。异常染色体核型描述根据2009年ISCN (International System for Human Cytogenetic Nomenclature) 标准[14] 和 IWGMC(International Working Group on MDS Cytogenetics)共识指南 [15]。 因骨髓增生减低本身会造成染色体核型分析成功 率低,本研究用至少 10 个中期分裂相替代原标准 中至少 20 个中期分裂相作为可分析的样本。≥ 2 个分裂相的相同的结构异常或染色体增加认为是 存在异常克隆。≥ 3 个分裂相相同的染色体缺失 认为是存在异常克隆。 1.4 诊断标准
阵发性睡眠性血红蛋白尿(PNH)为至少 3 次 Ham 实验阳性或两系糖基磷脂酰肌醇锚蛋白 (GPI-AP)克隆缺失 >20% ,伴溶血的临床症状和 实验室检查证据(如血红蛋白尿、结合珠蛋白降 低、乳酸脱氢酶增高及尿胆红素增高)。PNH 克 隆采用流式细胞技术检测(抗-CD55 和抗-CD59)。 MDS 及 AML 的诊断及分型根据 WHO 2008 标准 [1]。 1.5 随访
随访方式为每 3~6 月一次门诊或住院复诊时 评估。所有患儿均在2013年6~7月电话随访一 次最新情况。随访终点为患儿死亡或2013年7月 30日。最终有130例RCC患儿可随访到临床转 归。失访率14%,中位随访时间为36个月(范围 1~283个月)。随访评估内容包括全血细胞计数、 骨髓细胞形态学、骨髓组织病理学、染色体核型 分析及血细胞 CD55/CD59 表达水平等。 1.6 疗效评价标准
参考文献标准判定疗效等级(表 1)[16, 17]。必 须以至少连续两次、间隔至少 4 周全血细胞计数 结果来评估疗效。
| 表 1 非重型 AA 及 RCC 疗效标准 |
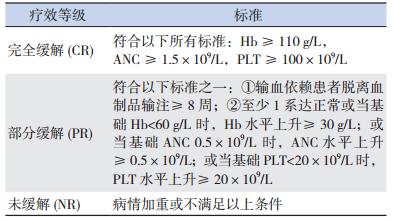
所有统计学处理均应用 SPSS 17.0 统计软件完 成。非正态分布资料用中位数(范围)表示,组 间比较采用 Mann-Whitney U检验。总体生存采用 Kaplan-Meier 法估算,组间比较采用非参数检验。 多因素分析均采用 Cox 回归模型。P<0.05为差异 有统计学意义。 2 结果 2.1 RCC 患儿与非重型 AA 患儿临床特征的比较
本研究共纳入 1 420 例既往诊断为非重型 AA 的患儿,最终经过骨髓细胞形态学和骨髓病理学 回顾性分析,152 例(10.70%)重新诊断为 RCC, 其余仍诊断为非重型 AA。RCC 患儿与非重型 AA 患儿临床特征见表 2。与非重型 AA 相比,RCC 患 儿发病时 Hb 较低(P=00.001),胎儿血红蛋白(fetal hemoglobin,HbF)较高(P=00.037)。
| 表 2 RCC 患儿与非重型 AA 患儿临床特征的比较 |
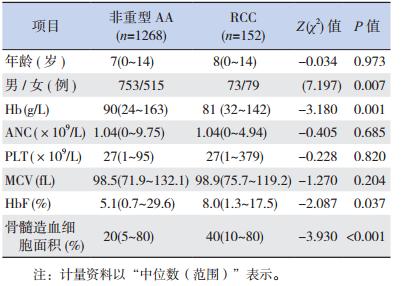
骨髓细胞形态学和组织病理学分析显示 RCC 患儿红系、粒系和巨核细胞病态造血的比例分别 为 75.0%、53.9% 和 40.1%(巨核细胞系病态造血 通过 CD41 免疫组化染色确定)。RCC 患儿 1 系、 2 系、3 系血细胞病态造血的比例分别为 44.7%、 36.2%、19.1%,而非重型 AA 患儿仅有 12.1% 表 现为单纯红系的轻度病态造血。RCC 造血细胞面 积高于非重型AA(P<0.001)。22.4% 和 31.0% 的 RCC 患儿可见淋巴样小巨核细胞和异常红细 胞造血岛。轻度的纤维化(3.5%)和不成熟前体 细 胞 异 常 定 位(abnormal localization of immature precursors,ALIP)(1.1%)在 RCC 患儿中罕见, 在非重型 AA 患儿未见。 2.2 细胞遗传学分析
79 例 RCC 和 302 例非重型 AA 有可供分析的 染色体核型结果。RCC 患儿发病时染色体核型异 常的比例高于非重型 AA[21.5%(17/79)vs 8.3% (25/302),P=00.001]。RCC 异常染色体核型包括: 7 号染色体单体 23.5%(4/17)、8 号染色体三体 17.6%(3/17)、复杂核型 17.6%(3/17)和其他异 常 41.2%(7/17)。有 4 例(4/62,6.5%)发病时 染色体核型正常的 RCC 患儿病程中出现异常染色 体核型,分别为 1 例 46,XY,i(7q),1 例两个克 隆 47,XY,+8,t(8;21)(q22;q22),-12, del(20)(q11),+21 和 46,XY,del(20)(q12) 和 2 例单纯 8 号染色体三体(表 3)。
| 表 3 克隆演变 RCC 患儿的临床特征 |

RCC患儿接受下列4种治疗方案之一: (1)仅观察及支持治疗(15.2%);(2)仅雄激 素(包括安雄、达那唑及司坦唑醇)(16.0%)治疗; (3)仅环孢素(CSA)治疗(15.2%); (4)CSA和雄激素联合用药(53.6%)。接受药 物治疗的RCC患儿总体CR、PR、NR率分别为 19.0%(20/105)、26.7%(28/105)、54.3%(57/105)。 3个不同药物治疗组间疗效比较差异无统计学意 义(P=00.849)。未经药物治疗的观察组和药物治 疗组间疗效比较差异无统计学意义(P=00.498)。 RCC 患儿 6 个月、12 个月、24 个月有效率(CR+PR) 分 别 为 16.7%(21/126)、26.0%(32/123)、 45.4%(44/97)。
6 例RCC 患儿在全血细胞计数进展至重型 AA/超重型AA水平时,接受了抗人T淋巴细胞免 疫球蛋白(ATG)联合CSA的强烈免疫抑制治疗 (IST),其中1例达CR,5例NR。3例患儿在 全血细胞计数进展至重型AA/超重型AA水平时 接受了造血干细胞移植。上述9例患儿未计入最 终的生存及白血病转化分析。 2.4 疗效预测因素
单因素分析表明患儿性别、年龄、诊断到开 始药物治疗的时间、治疗前 Hb、ANC、PLT 水平、 异常染色体核型、病态造血系别、骨髓造血细胞 面积与总 CR 率、总有效率均无相关性。多因素分 析未发现与总 CR 率、总有效率相关的独立因素。 2.5 复发
至随访终点,36.1%(44/122)RCC 患儿停药。 其中 25%(11/44)RCC 患儿停药后复发。除 1 例 患儿复发前有上呼吸道感染外,其余均无诱因。 停药后复发的中位时间为停药后 6.5 个月(范围 1~87 个月)。停药后复发的 11 例 RCC 患儿中, 3 例用原方案治疗仍有效。停药后复发与治疗方 案、CSA 疗程、雄激素疗程及 CSA 减量时间均无 关。Kaplan-Meier 法估计停药后 2 年累计复发率为 28.4%。11.5%(14/122)的患儿为治疗过程中复发 (即失去原来的 CR 或 PR),其中 3 例复发前有 上呼吸道感染,1 例伴 PNH,1 例伴染色体核型异 常,余均无诱因(表 3)。 2.6 总体生存及克隆演变分析
与非重型AA相比,RCC患儿总体生存较差 (P=00.032,图 1),5年和10年的预期生存率分 别为87.9%和72.4%,非重型AA分别为93.8%和 85.7%。本研究中克隆演变包括3种情况:急性白 血病、PNH和新出现的染色体异常,对非重型AA 患儿还包括MDS。至随访终点,RCC患儿克隆演 变率为10.0%(13/130)(表 3)。与非重型AA相比, RCC患儿克隆演变的风险较高(P<0.001,图 2), RCC患儿5年和10年的预期克隆演变率分别为 15.3%和20.0%,非重型AA患儿为0.8%和2.7%。 与非重型AA相比,RCC患儿病程中新出现染色 体核型异常的风险较高(P<0.001,图 3),RCC 患儿2年预期出现新的染色体异常率为3.6%,非 重型AA患儿为0.2%。RCC和非重型AA患儿10 年的预期PNH进展率分别为0.5%、0.3%,两者比 较差异无统计学意义(P=00.396)。RCC患儿5年 和10年预期白血病转化率为10.0%和20.0%,非 重型AA患儿为0.2%和1.2%,与非重型AA相比, RCC患儿白血病转化的风险较高(图 4)。
 |
图 1 非重型 AA 和 RCC 总体生存情况 与非重型 AA(NSAA)相比,RCC 患儿总体生存较差,5 年和 10 年的预期 生存率分别为 87.9% 和 72.4%,非重型 AA(NSAA)分别为 93.8% 和 85.7%。 |
 |
图 2 非重型 AA 和 RCC 总体总体克隆演变情况 与非重型 AA(NSAA)相比,RCC 患儿克隆演变的风险较高, RCC 患儿 5 年和 10 年的预期克隆演变率分别为 15.3% 和 20.0%, 非重型 AA(NSAA)患儿为 0.8% 和 2.7%。 |
 |
图 3 非重型 AA 和 RCC 出现新的核型异常的克隆演 变情况 与非重型 AA(NSAA)相比,RCC 患儿病程中新出现 染色体核型异常的风险较高,RCC 患儿 2 年预期出现新的染色体 异常率为 3.6%,非重型 AA(NSAA)患儿为 0.2%。 |
 |
图 4 非重型 AA 和 RCC 向白血病转化的克隆演变 情况 与非重型 AA(NSAA)相比,RCC 患儿白血病转化的 风险较高,RCC 患儿 5 年和 10 年预期白血病转化率为 10.0% 和 20.0%,非重型 AA(NSAA)患儿为 0.2% 和 1.2%。 |
单因素分析表明发病时具有下列因素与 RCC 患儿总体生存率不良相关:Hb<90 g/L(P=00.022)、 ANC<1.2×109/L(P=00.006)、 发 病 时 染 色 体 核 型异常(P=00.011)、不良核型(涉及 7 号染色 体的异常核型或复杂核型)(P=00.011)、治疗 前 HbF ≤ 0.025(P=00.003)、骨髓 PAS 染色阳性 (P=00.049)。接受不同药物治疗方案的 RCC 患儿 总体生存率和白血病转化率差异无统计学意义; 接受支持治疗与药物治疗 RC 患儿总体生存率和白 血病转化率差异亦无统计学意义。 3 讨论
RCC 是2008 年WHO 造血与淋巴系统肿瘤 诊断分型标准对儿童 MDS 暂定的分类。RCC 是 儿童 MDS 最常见的类型,约占儿童 MDS 患者的 50% [1]。目前,对于这一以形态学为基础诊断的类 型尚有争议,原因在于:既往研究报道 RCC 具有 与 AA 相似的临床特征、对 IST 的疗效和临床转归 亦相似[18, 19];一部分先天性骨髓衰竭与 RCC 骨髓 形态相似[20, 21];全基因组测序发现 RCC 和儿童非 重型 AA 均表现为突变基因较少,两者在单个样本 平均突变数和具体突变基因上无显著性差异[19]。 因此,这种基于形态学的疾病分类的意义受到了 质疑。既往研究着重于 RCC 与重型 AA/ 超重型 AA 的比较,且随访时间较短。大多 RCC 患儿骨 髓增生程度较重型 AA/ 超重型 AA 轻,全血细胞 计数水平更接近于非重型 AA。非重型 AA 和 RCC 均表现为慢性病程,随访时间足够长才能获得准 确的临床转归。
本研究发现 RCC 患儿外周血 Hb 水平较低, 但骨髓增生程度正常甚至较活跃,特别是红系增 生较活跃,表现为红系比例增高、异常红系造血 岛及显著红系病态造血,因此 RCC 患儿具有无效 造血的特点。既往报道的较高的骨髓造血细胞面 积与非重型 AA 预后良好有关不同[22],本研究中 较高的骨髓造血细胞面积与 RCC 患儿的疗效及总 体生存无关,提示骨髓衰竭程度与 RCC 预后无关。
RCC 患儿具有克隆性造血的特点。与既往报 道非重型 AA 患儿相比[18, 19, 22],RCC 患儿起病时染 色体核型异常率较高,随着病程进展,克隆演变 率亦较高,尤其是向恶性克隆转化的风险较高。 分析表明非重型 AA 中向白血病转化的患儿可能与 治疗相关,如长期应用 G-CSF,多次强烈 IST 及相 继接受 ATG 及 CTX 治疗等。而 RCC 患儿中转化 为白血病者与具有 7 号染色体异常或复杂染色体 核型有关,与治疗无关。提示 RCC 本身具有较高 的向白血病转化的风险。另一方面,RCC 患儿向 PNH 转化率低。既往研究认为 AA 患者出现 PNH 克隆演变的发病机制主要是免疫异常导致的在骨 髓衰竭背景上的 PNH 克隆细胞免疫逃逸[23, 24, 25, 26]。与 既往研究一致[27] ,本研究中部分 RCC 患儿对 IST 亦有效,提示部分患儿发病与免疫异常有关。上 述结果提示,依据形态学诊断的 RCC 虽然具有可 操作性,但对探寻疾病的本质尚存局限性。
既往研究结果表明,输血依赖和(或)粒细 胞小于 1.0×109/L 的 RCC 对 IST 疗效与重型 AA 或超重型 AA 相似[28]。本研究结果与之有所不同, RCC 患儿对以 CSA 为主免疫抑制剂反应较非重型 AA 差,完全缓解率低。进展至重型 AA 或超重型 AA 时,行 IST 的有效率亦显著低于既往报道。在 AA 患儿中,全血细胞计数、骨髓细胞造血面积均 与疗效显著相关[29],但上述因素均与 RCC 患儿疗 效无关。
既往研究认为 RCC 患儿临床转归与非重型 AA 患儿相似,甚至好于 AA [18, 30]。本研究长期随 访结果显示,RCC 患儿的总体生存较非重型AA 差。 可能原因有:(1)既往研究中主要以重型 AA/ 超 重型 AA 作为 RCC 的对照组,而大多数 RCC 的起 病时的病情与非重型 AA 相当,本研究也显示 RCC 起病时除 Hb 和 HbF 外,其他临床指标较非重型 AA 无显著性差异。因此,重型 AA/ 超重型 AA 本 身相对较差的预后可能导致结果偏倚。(2)既往 大多数研究随访时间较短,可能未随访至 RCC 克 隆演变时间。本研究发现 54% 克隆演变发生于诊 断后 24 个月,因此随访时间长更有利于观察到两 者远期预后的差异性。
综上所述,目前以形态学为基础诊断的 RCC 具有临床意义。RCC 具有 MDS 的特征,与非重型 AA 患儿相比,总体生存较差,恶性转化率较高, CSA 治疗无效率较高。同时,需强调目前诊断的 RCC 仍具有异质性,未来研究应着力寻找克隆性 证据,为从发病机制上鉴别两种疾病提供依据。
| [1] | Baumann I, Niemeyer CM, Bennett JM, et al. WHO classification of tumors of haematopoietic and lymphoid tissues[M]//Swerdlow SH, Campo E, Harris Nl, et al. Childhood Myelodysplastic Syndrome, 2008: 104-107. |
| [2] | Niemeyer CM, Baumann I. Myelodysplastic syndrome in children and adolescents[J]. Semin Hematol, 2008, 45(1): 60-70. |
| [3] | Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases[J]. Leukemia, 2003, 17(2): 277-282. |
| [4] | Kardos G, Baumann I, Passmore SJ, et al. Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7[J]. Blood, 2003, 102(6): 1997-2003. |
| [5] | Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study[J]. Blood, 1987, 70(6): 1718-1721. |
| [6] | 中国小儿血液与肿瘤杂志编辑委员会. 小儿再生障碍性贫血 诊疗建议[J]. 中国小儿血液与肿瘤杂志, 2007, 12(5): 236-240. |
| [7] | Yang W, Zhang P, Hama A, et al. Diagnosis of acquired bone marrow failure syndrome during childhood using the 2008 World Health Organization classification system[J]. Int J Hematol, 2012, 96(1): 34-38. |
| [8] | Baumann I, Führer M, Behrendt S, et al. Morphological differentiation of severe aplastic anaemia from hypocellular refractory cytopenia of childhood: reproducibility of histopathological diagnostic criteria[J]. Histopathology, 2012, 61(1): 10-17. |
| [9] | Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome[J]. Hematology Am Soc Hematol Educ Program, 2011, 2011: 84-89. |
| [10] | 杨文钰, 陈晓娟, 张培红, 等. 儿童血细胞减少伴骨髓增生减 低100 例临床特征分析[J]. 中国当代儿科杂志, 2013, 15(6): 448-452. |
| [11] | Bennett JM, Orazi A. Diagnostic criteria to distinguish hypocellular acute myeloid leukemia from hypocellular myelodysplastic syndromes and aplastic anemia: recommendations for a standardized approach[J]. Haematologica, 2009, 94(2): 264-268. |
| [12] | Cantù Rajnoldi A, Fenu S, Kerndrup G, et al. Evaluation of dysplastic features in myelodysplastic syndromes: experience from the morphology group of the European Working Group of MDS in Childhood (EWOG-MDS)[J]. Ann Hematol, 2005, 84(7): 429-433. |
| [13] | Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity[J]. Haematologica, 2005, 90(8): 1128-1132. |
| [14] | Shaffer LG, Slovak ML, Campbell LJ. An International System for Human Cytogenetic Nomenclature. Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature[M]. Basel, Switzerland: S. Karger AG, 2009. |
| [15] | Chun K, Hagemeijer A, Iqbal A, et al. Implementation of standardized international karyotype scoring practices is needed to provide uniform and systematic evaluation for patients with myelodysplastic syndrome using IPSS criteria: An International Working Group on MDS Cytogenetics Study[J]. Leuk Res, 2010, 34(2): 160-165. |
| [16] | Maciejewski JP, Sloand EM, Nunez O, et al. Recombinant humanized anti-IL-2 receptor antibody (daclizumab) produces responses in patients with moderate aplastic anemia[J]. Blood, 2003, 102(10): 3584-3586. |
| [17] | 张之南, 沈悌. 血液病诊断及疗效标准[M]. 第3 版. 北京: 科学出版社, 2007: 20-23. |
| [18] | Hama A, Muramatsu H, Ito M, et al. Risk factors for clonal evolution of acquired bone marrow failure after immunosuppressive therapy in children[J/OL]. Blood (ASH Annual Meeting Abstracts), 2013, 122(21): 2473. |
| [19] | Narita A, Muramatsu H, Yoshida K, et al. Whole exome sequencing shows a paucity of somatic gene mutations in pediatric idiopathic bone marrow failure syndrome[J/OL]. Blood (ASH Annual Meeting Abstracts), 2013, 122(21): 3708. |
| [20] | Yoshimi A, Niemeyer C, Baumann I, et al. High incidence of Fanconi anaemia in patients with a morphological picture consistent with refractory cytopenia of childhood[J]. Br J Haematol, 2013, 160(1): 109-111. |
| [21] | Karow A, Flotho C, Schneider M, et al. Mutations of the Shwachman-Bodian-Diamond syndrome gene in patients presenting with refractory cytopenia-do we have to screen[J]. Haematologica, 2010, 95(4): 689-690. |
| [22] | DeZern AE, Pu J, McDevitt MA, et al. Burst-forming uniterythroid assays to distinguish cellular bone marrow failure disorders[J]. Exp Hematol, 2013, 41(9): 808-816. |
| [23] | Maciejewski JP, Rivera C, Kook H, et al. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol-anchored protein-deficient clones[J]. Br J Haematol, 2001, 115(4): 1015-1022. |
| [24] | Wang H, Chuhjo T, Yasue S, et al. Clinical significance of a minor population of paroxysmal nocturnal hemoglobinuria-type cells in bone marrow failure syndrome[J]. Blood, 2002, 100(12): 3897-3902. |
| [25] | Dunn DE, Tanawattanacharoen P, Boccuni P, et al. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes[J]. Ann Intern Med, 1999, 131(6): 401-408. |
| [26] | Sugimori C, Chuhjo T, Feng X, et al. Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia[J]. Blood, 2006, 107(4): 1308-1314. |
| [27] | Hata T, Tsushima H, Baba M, et al. Long-term outcome of immunosuppressive therapy for Japanese patients with lowerrisk myelodysplastic syndromes[J]. Int J Hematol, 2013, 98(6): 687-693. |
| [28] | Yoshimi A, van den Heuvel-Eibrink MM, Baumann I, et al. Comparison of horse and rabbit antithymocyte globulin in immunosuppressive therapy for refractory cytopenia of childhood[J]. Haematologica, 2014, 99(4): 656-663. |
| [29] | Yoshida N, Yagasaki H, Hama A, et al. Predicting response to immuno-suppressive therapy in childhood aplastic anemia[J]. Haematologica, 2011, 96(5): 771-774. |
| [30] | Hama S, Muramatsu H, Ito M, et al. Comparison of clinical outcome between children with aplastic anemia and refractory cytopenia of childhood who received immunosuppressive therapy with anti-thymocyte globulin and cyclosporine[J/OL]. Blood (ASH Annual Meeting Abstracts), 2011, 118: 53. |