 2015, Vol. 17
2015, Vol. 17



急性髓系白血病(acute myeloid leukemia, AML)是一组在临床及细胞遗传学上均具有高度 异质性的疾病,具有多种遗传性缺陷[1]。初诊时的 细胞遗传学(染色体核型分析)检测结果已经作为AML 的重要的预后因素[2]。研究发现,除了遗 传学的改变之外,表观遗传学在肿瘤发生的过程 中也起到了重要作用[3]。越来越多的研究表明, DNA 甲基化状态异常尤其是抑癌基因的高甲基化 失表达在白血病的发生发展、诊断、检测微小残 留病、预测病情以及指导治疗方面都有重要的作 用[3, 4, 5, 6, 7, 8]。
9 号染色体短臂21 段(9P21)的异常在多 种肿瘤的发生发展过程中起着重要的作用。目前 已经发现在9P21 区段有P14/ARF、P16/INK4A、 P15/INK4B 等抑癌基因,它们的位置非常临近,并 且有部分重叠,构成了一个重要的抑癌基因簇。 通过抑制细胞周期蛋白依赖性激酶4 和6(cyclindependent kinases 4 and 6) 的活性,使细胞周期 停滞于G1/S 期,对细胞周期起着负性调控作用 [9, 10]。范丽萍等[11] 采用巢式甲基化特异性聚合酶 链反应法观察了82 例急性白血病患者中P16 基 因甲基化,结果显示P16 基因高甲基化发生率为 39.0%; 在成人AML 中,有50%~80% 的患者可 以检测到CDKN2B 基因(P15) 甲基化[8, 12]。关 于AML 中基因甲基化的研究在国内外已经较多, 然而多是成人AML 的研究,在儿童AML 中的研 究相对较少。为了解我国儿童AML 中P14/ARF、 P16/INK4A、P15/INK4B 基因甲基化的情况,本研 究应用甲基化特异性- 多重连接酶依赖性探针扩增 法(methylation-specific multiplex ligation-dependent probe amplification,MS-MLPA) 对58 例初诊AML 患儿进行9P21 区段P14/ARF、P16/INK4A、 P15/INK4B 基因甲基化的研究,并分析其与临床特 征、疗效及预后的相关性,报告如下。 1 资料与方法 1.1 研究对象
收集2010 年4 月至2012 年12 月我院儿科收 治的58 例初诊为AML 的住院患儿的临床资料并 进行回顾性分析,包括临床特征、FAB 分型及细 胞遗传学改变等。所有患儿均经骨髓形态学、细 胞组织化学染色、细胞免疫分型及细胞遗传学确 诊。另选取38 例健康志愿儿童作为正常对照。本 研究获得AML 患儿及健康儿童志愿者监护人的知 情同意。 1.2 治疗方案
(1)急性早幼粒细胞白血病(APL,AMLM3) 患儿治疗方案: 应用全反式维甲酸 (ATRA,每日25 mg/m2,连续使用42 d)联合三 氧化二砷(As2O3,每日0.15~0.2 mg/kg,连续使 用28 d)行诱导治疗达完全缓解(CR)后,给予 去甲氧柔红霉素(IDA,每日10 mg/m2,连续使用 3 d),再予As2O3(每日0.15~0.2 mg/kg,连续使 用28 d)行强化治疗。上述治疗结束后随机分为 两组,分别予柔红霉素(DNR,每日45 mg/m2) 或DNR( 每日45 mg/m2)+ 阿糖胞苷(Ara-C, 每12 h 1 000 mg/m2),每疗程均连续使用3 d, 连用2 个疗程。维持治疗期采用ATRA( 每日 20 mg/m2,1~14 d) 联合6- 巯基嘌呤(6-Mp,每 日100 mg/m2,15~28 d) 和甲氨蝶呤(MTX,每 日20 mg/m2,15 d 和22 d)进行治疗,共维持治 疗2 年。缓解期行腰穿和地塞米松(4 mg)+Ara-C (36 mg)+MTX(12 mg)的三联鞘注4 次。
(2)非AML-M3 患儿治疗方案:该组患儿采 用AML-99 方案进行化疗,详见文献[13]。 1.3 基因组DNA 提取
取AML 患儿和健康儿童志愿者骨髓液或 外周血2 mL,采用DNA 提取试剂盒(Axygen Biosciences 公司,美国)提取基因组DNA,-20℃ 保存备用。 1.4 MS-MLPA 法行甲基化检测
所用试剂盒购于荷兰MRC-Holland 公司 (ME024-B1),具体操作步骤如下:(1)DNA 变性: 取5 μL(20 ng/μL)DNA 样品,98 ℃ 处理 5 min,25℃暂停。(2)探针与DNA 的杂交:取 出样品,降至室温,加入3 μL 杂交混合液(1.5 μL SALSA probemix+1.5 μL MLPA 缓冲液),95℃处 理1 min,60℃温浴16~24 h。(3)连接及连接- 消化:PCR 仪降到20℃,取出PCR 管;每管加 入3 μL ligase buffer A 和10 μL 水,吹打混匀;每 个标本分为a、b 两管,各取10 μL 混合液到新的 PCR 管;将所有管放回PCR 仪,48℃暂停;往a 管中加入10 μL 连接酶混合液(8.25 μL 水+1.5 μL ligase buffer B+0.25 μL ligase-65 酶),混匀,往 b 管中加入10 μL 连接酶- 消化混合液(7.75 μL 水+1.5 μL ligase buffer B+0.25 μL ligase-65 酶+0.5 μL HhaI 酶),混匀;48℃温浴30 min 连接消化;98 ℃ 加热5 min 灭活酶;20 ℃ 暂停。 (4)接探针的PCR 扩增:从PCR 仪中取出样品, 降至室温;加入10 μL PCR 混合液(7.5 μL 水+2 μL PCR Primer Mix+0.5 μL SALSA Polymerse) 行PCR 扩增,95℃变性30 s,60℃退火30 s,72℃延伸 60 s,35 个循环后,72℃再延伸20 min,15℃暂停。 (5)PCR 产物的毛细管电泳:取0.5 μL PCR 产物, 加入0.5 μL 500LIZ® Size Standard 和9 μL Hi-DiTM Formamide 行毛细管电泳。(6)结果分析:所得 结果通过GeneMapper 4.0 和Coffalyser 软件进行半 定量分析,了解目的基因的片段缺失或扩增情况。
该MS-MLPA 试剂盒中,包含9 个甲基化检测 探针,可检测CDKN2A(P14/ARF、P16/INK4A)、 CDKN2B(P15/INK4B)基因启动子区域的甲基化 状态。各探针的详细情况见表 1。
| 表 1 MS-MLPA 试剂盒中甲基化特异性探针列表 |

随访时间截止至2013 年12 月20 日或失访日 期,中位随访时间为16 个月(1~45 个月)。无事 件生存(EFS)期定义为自诊断到第1 次事故(包 括分子生物学和临床复发、在CR 期间的死亡)或 末次随访日期;无病生存(DFS)期定义为自CR 至白血病复发或在CR 期间死亡或末次随访日期。 总生存(OS)期指自诊断到死亡或末次随访日期。 1.6 统计学分析
采用SPSS 16.0 统计软件对数据进行统计学分 析,计量资料以中位数(范围)表示,组间比较采 用秩和检验;计数资料用百分率(%)表示,样本 率的比较采用χ2 检验。生存分析应用Kaplan-Meier 法和log-rank 检验。p<0.05 为差异有统计学意义。 2 结果 2.1 AML 患儿的一般资料
58 例AML 患儿中,男36 例(62%),女 22 例(38%),中位年龄8 岁(1~13 岁)。中位 初诊白细胞数为17.3×109/L(0.8~504.8×109/L), 中位初诊血红蛋白为85 g/L(43~134 g/L),中位 初诊血小板数为44×109/L(9~228×109/L)。 2.2 MS-MLPA 法行甲基化检测结果
在健康儿童中各探针均未发现甲基化。在 AML 患儿中,157 bp 探针、427 bp 探针和171 bp 探针未检测到甲基化;检测到CDKN2A 基因的甲 基化包含136 bp 探针和237 bp 探针; CDKN2B 基 因的甲基化涉及130 bp 探针、210 bp 探针、220 bp 探针和417 bp 探针。 2.2.1 甲基化特异性探针检测结果与临床特征、预后的关系
在44 例(76%)AML 患儿中检测到 至少1 个探针的甲基化,37 例(64%)AML 患儿 中检测到至少2 个探针的甲基化。依据每一个探 针的检测结果分为非甲基化组和甲基化组。各探 针非甲基化组和甲基化组之间进行比较后发现, 136 bp 探针甲基化组患儿的血红蛋白水平[123.5 g/L (113~134 g/L)] 高于非甲基化组[84 g/L(43~127 g/L)] (p=0.026),130 bp 探针甲基化组患儿的血小板 水平[38×109/L(9~160×109/L)] 低于非甲基化组 [75×109/L(30~228)×109/L](p=0.029),417 bp 探针甲基化组患儿的男性比例高于非甲基化组 (p=0.003)。不同FAB 分型AML 患儿各探针甲基 化百分比情况见表 2,只有130 bp 探针在不同FAB 分型患儿中甲基化的比例有明显的差异(p=0.034)。 各探针的非甲基化组和甲基化组的EFS、DFS 和OS 率之间差异均无统计学意义,然而220 bp 探针甲基 化组的DFS 和EFS 率相比非甲基化组有降低趋势。 患者年龄与患者甲基化探针数目之间无明显的相 关性;患儿高甲基化的探针数目与其EFS、DFS 及 OS 率之间无明显的相关性。
| 表 2 不同FAB 分型AML 患儿CDKN2A 与CDKN2B 基因甲基化发生率 [例(%)] |
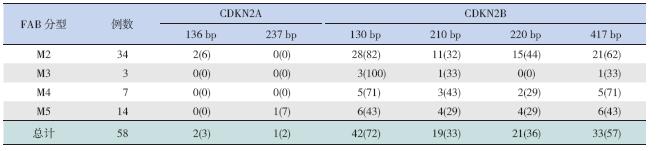
AML 患儿中, CDKN2A 的甲基化率仅为5%,而CDKN2B 的甲 基化较为常见,各探针甲基化率为76%。AML-M3 和AML-M4 的CDKN2B 甲基化发生率为100%。 随访结束时3 例AML-M3 患儿均无病生存;7 例 AML-M4 患儿中,4 例为M4eo(CBFB/MYH11 阳性), 1 例放弃治疗,3 例复发。 2.2.3 基因甲基化与细胞遗传学结果之间的关系
46 例患儿进行细胞遗传学检测,1 例未见分裂 像。按照文献[2] 所述,依据细胞遗传学结果将45 例患儿分为预后良好组(n=27),其中t(8;21) 21 例,t(15;17)3 例,inv16(CBFB/MYH11)3 例; 预后中等组(n=11)和预后不良组(n=7)。上述 3 组中均有基因甲基化,预后良好组患儿发生甲 基化24 例(89%),预后中等组患儿发生甲基化 7 例(64%),预后不良组患儿发生甲基化5 例 (71%),三组间比较差异无统计学意义(p=0.174)。 3 讨论
AML 是一种克隆性起源于髓系多能干细胞 或很早期的祖细胞突变而引起的造血系统恶性肿 瘤。近半数AML 患者存在细胞遗传学异常,细胞 遗传学正常的患者,可存在其他的体细胞突变, 影响患者的预后[14]。除了遗传学改变之外,表观 遗传学在肿瘤发生中也起到了重要的作用[3]。基 因的甲基化在表观遗传学的研究中也越来越引起 重视。富含CG 的区域,即CpG 岛多位于基因启 动子区,并且一般在健康组织中为非甲基化[4, 15, 16] 状态。这些基因的启动子区的甲基化常会导致基 因的转录沉默,是肿瘤发生过程中基因沉默的重 要机制之一[4, 12]。其中最常研究的是CDKN2B 基 因(P15/INK4B)和与它功能类似的CDKN2A 基 因(包含很多转录变体:亚型1,P16/INK4A;亚 型4,P14/ARF)[17]。本研究应用MS-MLPA 方法 对58 例初诊儿童AML 的9P21 区域的CDKN2A 和 CDKN2B 基因进行甲基化检测,发现CDKN2A 的 甲基化率仅为5%,而CDKN2B 的甲基化较为常见, 占76%。
Galm 等[7] 的研究认为基因甲基化与OS 及初 诊时的白细胞计数无关。而本研究中发现,部分 探针甲基化与初诊时的性别、白细胞和血小板计 数有关。其原因可能与不同的实验方法、不同年 龄阶段等有关,需要进一步扩大样本量进行深入 研究。
有研究认为,约50%~60% 的APL 患者发生 P15 启动子区的甲基化,并且在未联合应用As2O3 时,P15 启动子区的甲基化为预后不良的因素[18]。 当复发APL 患者应用As2O3 治疗之后,P15 甲基化 不影响预后[19]。在本研究的APL 患者中,P15 甲 基化率为100%,与既往报道文献并不完全一致, 可能与不同的检测方法、年龄及样本量小有关。 同时,本研究中的3 例P15 甲基化患儿均长期无 病生存,可能与应用含有As2O3 的治疗方案有关。 相似的,Markus 等[20] 的研究中,inv(16)的P15 平均甲基化水平为4.6%(2%~11%),远低于本 研究中AML-M4 患儿中P15 甲基化率(100%), 亦可能与不同的检测方法、年龄差别有关。
Cechova 等[12] 也应用了MS-MLPA 方法检测 9P21 区域甲基化的情况,其中13 例成人AML 的 P15 的甲基化率为100%,P14 甲基化率为62%, P16 甲基化率为46%。国内范丽萍等[11] 研究成 人AML,P16 基因甲基化率为39%。本研究中, P15 甲基化率与成人的相似,而P14 和P16 的甲 基化率则明显低于成人,其原因可能与研究样本 的年龄差异有关。有研究表明,在AML 中存在 IDH1/2、DNMT3A、TET2 等基因的重现性突变, 这些基因编码的蛋白质参与了表观遗传学途径, 破坏胞嘧啶共价修饰的平衡可能是髓系白血病发 病的关键步骤[4, 5]。然而,众所周知,儿童AML 中IDH1/2、DNMT3A、TET2 的突变率极低,与成人 存在明显差异[21, 22]。这提示,儿童AML 的发病可 能存在其他机制。基于成人与儿童具有相似的P15 甲基化发生率,同时,在MDS 中,P15 甲基化的 发生率会随着疾病进展到AML 而明显增加[12, 23], 因而我们推测:通过对P15 甲基化相关的通路进 行研究可能发现儿童与成人AML 的共同发病机制。
众所周知,DNA 甲基化在AML 的发病中起 到一定的作用,而DNA 的甲基化过程是可逆的, DNA 甲基化转移酶是甲基化过程中关键的一环。 已有文献表明,DNA 甲基化转移酶抑制剂在AML 的治疗中是安全有效的[24]。遗憾的是本研究中的 病例尚无应用该类药物的经验。此外,本组患儿 不同FAB 分型组别间样本含量差别较大,如M3 型组仅3 例,且总体样本量偏小,可能对结果有 一定的影响,需要进一步扩大样本量进行深入研 究。加强儿童AML 中基因甲基化的基础和临床方 面的研究是我们努力的方向。
| [1] | Caligiuri MA, Strout MP, Gilliland DG. Molecular biology of acute myeloid leukemia[J]. Semin Oncol, 1997, 24(1): 32-44. |
| [2] | Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties[J]. Blood, 1998, 92(7): 2322-2333. |
| [3] | 李玉华, 文飞球. DNA 甲基化与儿童白血病的临床研究进展[J]. 中国当代儿科杂志, 2011, 13(2): 174-177. |
| [4] | Schoofs T, Berdel WE, Müller-Tidow C. Origins of aberrant DNA methylation in acute myeloid leukemia[J]. Leukemia, 2014, 28(1): 1-14. |
| [5] | Jasielec J, Saloura V, Godley LA. The mechanistic role of DNA methylation in myeloid leukemogenesis[J]. Leukemia, 2014, 28(9): 1765-1773. |
| [6] | Das PM, Singal R. DNA methylation and cancer[J]. J Clin Oncol, 2004, 22(22): 4632-4642. |
| [7] | Galm O, Wilop S, Lüders C, et al. Clinical implications of aberrant DNA methylation patterns in acute myelogenous leukemia[J]. Ann Hematol, 2005, 84 Suppl 1: 39-46. |
| [8] | Agrawal S, Unterberg M, Koschmieder S, et al. DNA methylation of tumor suppressor genes in clinical remission predicts the relapse risk in acute myeloid leukemia[J]. Cancer Res, 2007, 67(3): 1370-1377. |
| [9] | Recio JA, Noonan FP, Takayama H, et al. Ink4a/arf deficiency promotes ultraviolet radiation-induced melanomagenesis[J]. Cancer Res, 2002, 62(22): 6724-6730. |
| [10] | Bazan V, Zanna I, Migliavacca M, et al. Prognostic significance of p16INK4a alterations and 9p21 loss of heterozygosity in locally advanced laryngeal squamous cell carcinoma[J]. J Cell Physiol, 2002, 192(3): 286-293. |
| [11] | 范丽萍, 沈建箴, 叶宝国, 等. 巢式MSP 法检测急性白血病 患者p16 基因甲基化状态[J]. 中国实验血液学杂志, 2007, 15(2): 258-261. |
| [12] | Cechova H, Lassuthova P, Novakova L, et al. Monitoring of methylation changes in 9p21 region in patients with myelodysplastic syndromes and acute myeloid leukemia[J]. Neoplasma, 2012, 59(2): 168-174. |
| [13] | Tsukimoto I, Tawa A, Horibe K, et al. Risk-stratified therapy and the intensive use of cytarabine improves the outcome in childhood acute myeloid leukemia: the AML99 trial from the Japanese Childhood AML Cooperative Study Group[J]. J Clin Oncol, 2009, 27(24): 4007-4013. |
| [14] | Mrózek K, Marcucci G, Paschka P, et al. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification?[J]. Blood, 2007, 109(2): 431-448. |
| [15] | Caiafa P, Zampieri M. DNA methylation and chromatin structure: the puzzling CpG islands[J]. J Cell Biochem, 2005, 94(2): 257-265. |
| [16] | Herman JG, Graff JR, Myöhänen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands[J]. Proc Natl Acad Sci U S A, 1996, 93(18): 9821-9826. |
| [17] | Tien HF, Tang JH, Tsay W, et al. Methylation of the p15(INK4B) gene in myelodysplastic syndrome: it can be detected early at diagnosis or during disease progression and is highly associated with leukaemic transformation[J]. Br J Haematol, 2001, 112(1): 148-154. |
| [18] | Chim CS, Kwong YL. Adverse prognostic impact of CDKN2B hyper-methylation in acute promyelocytic leukemia[J]. Leuk Lymphoma, 2006, 47(5): 815-825. |
| [19] | Au WY, Fung AT, Ma ES, et al. Serial studies of methylation of CDKN2B and CDKN2A in relapsed acute promyelocytic leukaemia treated with arsenic trioxide[J]. Br J Haematol, 2005, 131(5): 632-635. |
| [20] | Markus J, Garin MT, Bies J, et al. Methylation-independent silencing of the tumor suppressor INK4b (p15) by CBFbeta-SMMHC in acute myelogenous leukemia with inv(16)[J]. Cancer Res, 2007, 67(3): 992-1000. |
| [21] | 周剑锋, 张丽, 曾慧敏, 等. 儿童急性髓系白血病DNMT3a 基因突变的分析[J]. 中国实验血液学杂志, 2012, 20(6): 1297-1301. |
| [22] | 张丽, 曾慧敏, 艾晓非, 等. 儿童急性髓系白血病NPMl 和 IDH 基因突变的研究[J]. 中华血液学杂志, 2013, 34(5): 449-452. |
| [23] | 任媛媛, 竺晓凡. 儿童骨髓增生异常综合征分子生物学研究 进展[J]. 中国当代儿科杂志, 2014, 16(9): 957-961. |
| [24] | Pleyer L, Burgstaller S, Girschikofsky M, et al. Azacitidine in 302 patients with WHO-defined acute myeloid leukemia: results from the Austrian Azacitidine Registry of the AGMT-Study Group[J]. Ann Hematol, 2014, 93(11): 1825-1838. |