 2015, Vol. 17
2015, Vol. 17



代谢综合征在国内儿童中的发病率为1.4%~2.4%,在肥胖儿童中高达16.8%~28.8%[1]。其发病机制很复杂,研究表明,低出生体重儿或宫内生长迟缓(intrauterine growth retardation,IUGR)成年后发展成高血糖、高血脂、高血压的风险显著增加,因此,IUGR被认为是代谢综合征发病的危险因素之一[2]。随着社会发展、饮食结构和生活环境发生了较大的改变,IUGR后代更容易暴露于高脂饮食环境中。研究表明,在高脂饮食条件下,IUGR后代更容易出现胰岛素抵抗和高脂血症等代谢紊乱[3, 4]。然而,IUGR和高脂饮食对肝脏脂代谢相关基因转录水平的广泛调节改变尚无研究报道。本研究采用孕期全能量限制的方法建立IUGR模型,并在断奶后用高脂饲料喂养,研究IUGR和高脂饮食对子代体重、血脂及肝脏组织病理的联合作用,同时检测非酒精性脂肪肝(non-alcoholic fatty liver diseases,NAFLD)发病过程中多个关键基因的转录水平(涉及脂肪酸合成和氧化、胆固醇合成、转化和分泌等过程)[5],以了解其调节机制。
1 材料与方法 1.1 动物模型的建立采用孕期全程限食法建立IUGR大鼠模型,这是较常见并受认可的IUGR模型[6]。选取未曾交配的雄性和雌性Sprague-Dawley(SD)大鼠(250~300 g,购于北京维通利华实验动物技术有限公司),置于浙江大学实验动物中心四层(SPF级)喂养,室内环境条件保持相对稳定(室温:23±2℃;湿度:50%±10%;昼夜时间:12 h:12 h)。经过1周的适应性喂养,以雄雌1:2的比例进行交配,妊娠第0天定义为阴道涂片中出现精子当天。雌鼠怀孕后,随机分为正常组和限食组(摄食量为正常组的30%)。出生后,所有母鼠均不限制饮食,且新生鼠由自己的母亲喂养。为避免哺乳条件不同造成的差异,生后即将每笼子鼠数量调整为8只。
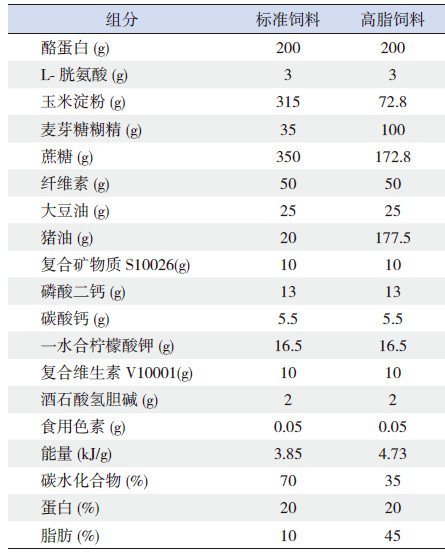
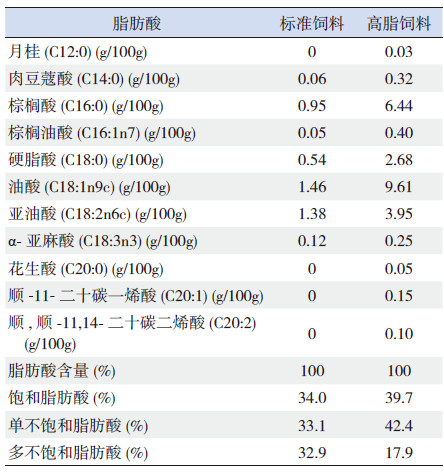
考虑到性别和性激素对脂代谢的影响,本研究验只采用雄性子鼠进行后续实验。于第21天断奶后,每笼取2只雄性子鼠,随机分配到标准饮食组(10%脂肪)和高脂饮食组(45%脂肪)。因此,实验对象共有4组:正常子鼠给予标准饮食(n=16)、正常子鼠给予高脂饮食(n=16)、IUGR子鼠给予标准饮食(n=12)和IUGR子鼠给予高脂饮食(n=12)。子鼠饲料均为纯化饲料(购于江苏美迪森生物医药有限公司),详细配方见表 1。此外,采用高效液相色谱法检测饲料中各类脂肪酸的成分(表 2)。在生后第70天,经过12 h禁食,用血糖仪测量鼠尾静脉血糖值后,腹腔内注射10%水合氯醛麻醉,采集血液和肝脏样本。
| 表 1 实验动物纯化饲料配方 |
| 表 2 实验动物纯化饲料的各类脂肪酸含量 |
出生时和出生后每周于清晨测量一次子鼠体重。断奶后,于每日清晨供给新鲜的饲料,并于次日晨丢弃剩余饲料,重新添加新鲜饲料。食物摄入量每周测定一次,定义为(供给饲料量-剩余饲料量)×100/体重,即为每日每100 g体重的进食量。能量摄入值为食物摄入量×饲料能量值。
1.3 血脂测量和肝脏组织病理学检测将采集的血液在室温下静置1 h后,以3000 rpm的速度离心5 min,收集上清。使用总胆固醇(total cholesterol,TC)和甘油三酯(triglycerides,TG)诊断试剂盒(南京建成生物工程研究所)测定样本的血脂水平。将新鲜肝脏组织在10%福尔马林中固定后,石蜡包埋,切成5 µm厚的薄片。经过苏木精-伊红(hematoxylin-eosin,HE)染色后,显微镜下观察肝脏组织切片。
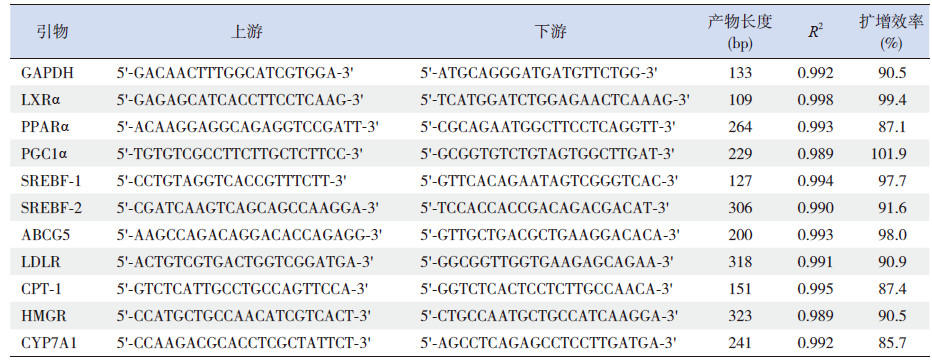
1.4 实时荧光定量PCR使用TRIzol(Life Technologies)抽提肝脏组织总RNA,并用NanoDrop 2000超微量分光光度计测定总RNA的浓度和纯度。鉴定合格后,取总RNA样本1 µg,使用高容量cDNA逆转录试剂盒(Applied Biosystems)进行逆转录得到单链cDNA。在罗氏LightCycler480 II实时荧光定量PCR系统上进行扩增反应。为了校正扩增效率对实验结果的影响,对每个基因均采用标准曲线来计算扩增效率,并使用分析软件对结果进行校正。分析目的基因时,采用管家基因甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)作为内参基因。本实验中采用的引物序列、扩增效率和产物长度见表 3。
| 表 3 引物序列、产物长度以及扩增效率 |
采用SPSS 22.0统计软件包进行数据处理,以P < 0.05为差异有统计学意义。数据以均数±标准差(x±s)表示。由于血清TG值呈偏态分布,在分析前先进行对数转换。两组间均数比较采用独立样本t检验;构成比(%)的比较采用卡方检验。各组间体重和摄入量的比较采用重复测量资料方差分析,事后检验采用LSD法。IUGR和高脂饮食的效应分析采用全因子模型的双因素方差分析。
2 结果 2.1 出生特征、生后体重改变和食物、能量摄入量出生时,正常雄性子鼠的体重为6.7±0.7 g(n=86),IUGR子鼠为4.3±0.6 g(n=88),两者差异有统计学意义(P < 0.01),且IUGR的平均体重低于正常子鼠的第5百分位。从出生后到3周龄,IUGR子鼠并未出现生长追赶的现象(图 1)。如图 2所示,重复测量资料方差分析结果显示,生后3~10周,高脂饮食下的IUGR大鼠出现了显著的生长追赶(其体重与标准饮食正常子鼠比较:P=0.392,与高脂饮食正常子鼠比较:P=0.191),而标准饮食的IUGR大鼠未出现显著的生长追赶。截止到10周,高脂饮食的正常大鼠并未出现显著的体重增加(与标准饮食正常子鼠比较:P=0.620)。
 |
图 1 两组子鼠生后0~3周的生长曲线 a示与正常子鼠比较,P < 0.01(重复测量资料方差分析)。 |
 |
图 2 4组子鼠生后3~10周的生长曲线 重复测量资料方差分析结果示4组间生后3~10周体重差异有统计学意义(P < 0.01),两两比较结果示标准饮食的正常子鼠、高脂饮食的正常子鼠和高脂饮食的IUGR子鼠之间体重差异无统计学意义,而标准饮食的IUGR子鼠的体重低于其他3组(P < 0.01)。a示与其他3组比较,P < 0.01。 |
重复测量资料双因素方差分析显示,IUGR显著增加食物摄入量(P < 0.001),而高脂饮食则降低食物摄入量(P < 0.001)。然而,由于高脂饲料的能量含量更高,我们采用能量摄入量来反映摄入水平。分析结果显示,与食物摄入量类似,IUGR大鼠有更高的能量摄入(P < 0.001),然而不同饮食组之间的能量摄入则差异无统计学意义(P=0.769)。从第4周开始,4组大鼠的能量摄入量均逐渐下降(图 3)。
 |
图 3 4组大鼠生后3~9周的每日能量摄入量 重复测量资料双因素方差分析结果示IUGR大鼠有更高的能量摄入(P < 0.001),而不同饮食组之间的能量摄入差异无统计学意义(P=0.769)。a示各组与本组前1周相比较,P < 0.05。 |
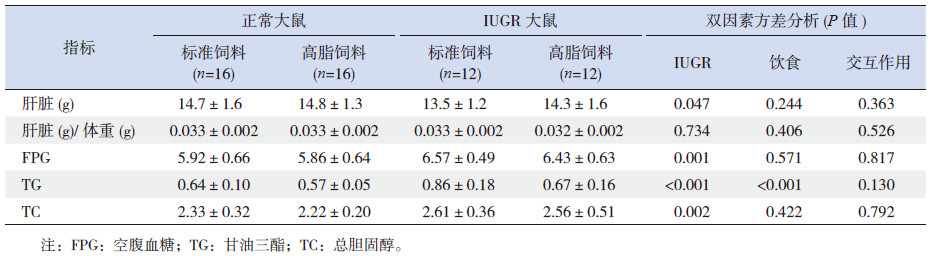
如表 4所示,IUGR大鼠与正常大鼠相比,空腹血糖(fasting plasma glucose,FPG)、TC和TG水平的差异有统计学意义。高脂饮食与标准饮食比较,FPG、TC差异无统计学意义,而血清TG水平的差异有统计学意义。 双因素方差分析显示IUGR和高脂饮食对FPG、TC和TG无交互作用(P>0.05)。IUGR显著减少肝脏重量(P=0.047),然而,在校正体重后(肝脏重量/体重),这种差异并不存在。
| 表 4 肝脏重量和血清生化指标(x±s ) |
如图 4所示,标准饮食条件下,相对于正常子鼠,IUGR大鼠的肝脏呈现散发的小泡性脂肪变性。在高脂饮食条件下,两组大鼠均出现脂肪堆积,表现为广泛的大泡性和小泡性脂肪变性,且IUGR大鼠的脂肪堆积更为严重。
 |
图 4 4组大鼠在10周时的肝脏组织病理切片 (HE染色,×400)A为标准饮食的正常大鼠;B为标准饮食的IUGR大鼠;C为高脂饮食的正常大鼠;D为高脂饮食的IUGR大鼠。标准饮食条件下,相对于正常子鼠,IUGR大鼠的肝脏呈现散发的小泡性脂肪变性;在高脂饮食条件下,两组大鼠均出现大泡性和小泡性脂肪变性(箭头所示),IUGR大鼠更为严重。 |
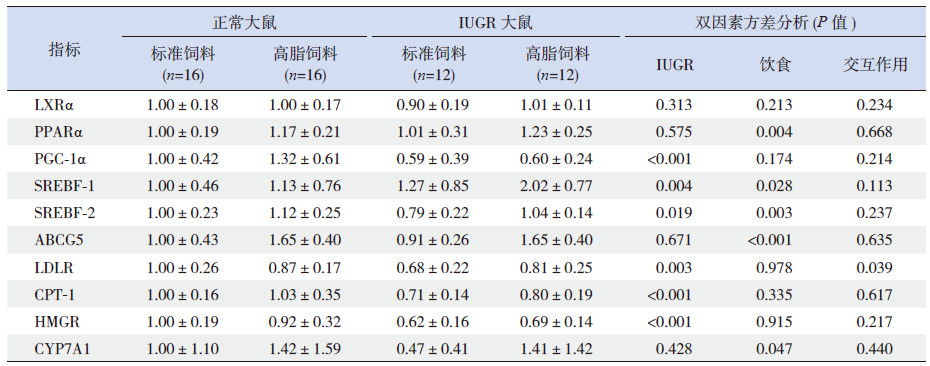
如表 5所示,IUGR大鼠与正常大鼠比较,过氧化物酶体增殖活化受体γ共激活因子-1α(peroxisome proliferator-activated receptor gamma coactivator 1-alpha,PGC-1α)、肉毒碱棕榈酰转移酶-1(carnitinepalmitoyltransferase I,CPT-1)、固醇调节元件结合因子-2(sterol regulatory element binding factor 2,SREBF-2)、3-羟基-3-甲基戊二酸单酰辅酶A还原酶(3-hydroxy-3-methylglutaryl-CoA reductase,HMGR)和SREBF-1的表达差异有统计学意义。高脂饮食与标准饮食比较,过氧化物酶体增殖物活化受体-α(peroxisome proliferator-activated receptor α,PPARα)、SREBF-1、SREBF-2、腺苷三磷酸结合盒转运体G5(ATP-binding cassette,sub-family G,member 5,ABCG5)和胆固醇7α-羟化酶(cholesterol 7 alpha-hydroxylase,CYP7A1)的表达差异有统计学意义。IUGR大鼠与正常大鼠比较,低密度脂蛋白受体(low density lipoprotein receptor,LDLR)水平差异有统计学意义,此外,IUGR和高脂饮食对LDLR有交互作用(P=0.039)。
| 表 5 大鼠肝脏相关基因的转录水平(根据2-△△ Ct法计算;x±s |
在哺乳期间给予过量营养后,IUGR大鼠会表现出生长追赶,并出现高血糖、高血脂及胰岛素抵抗等代谢紊乱[7, 8]。本研究发现,即使未出现生长追赶,IUGR大鼠也表现出明显的糖脂代谢紊乱。这与先前Lim等[9]的研究结果相似,他们的研究表明,相对于正常大鼠和出现生长追赶的IUGR大鼠,无生长追赶的IUGR大鼠反而更容易出现糖耐量异常。虽然标准饲料喂养的IUGR大鼠未出现生长追赶,但本研究发现,在高脂饮食条件下,IUGR大鼠出现了显著的生长追赶,而这种显著的体重增加并未在高脂饲料喂养的正常大鼠中出现。这说明在生命早期,IUGR大鼠更容易受到高脂饮食的影响,进而出现肥胖等表现。
虽然IUGR作为血糖和血脂异常的危险因素已被广泛认可,但其机制还未完全清楚。研究表明,IUGR大鼠的下丘脑食欲调节出现异常,导致摄入增加,进一步引起肥胖[10]。这与本研究中IUGR大鼠出现能量摄入增加的现象一致。此外,瘦素抵抗可能也参与了IUGR的食欲调控[11]。然而,食欲增强并不是IUGR引起肥胖和代谢紊乱的唯一原因。本研究发现,IUGR大鼠肝脏中PGC-1α和CPT-1的表达显著下调。CPT-1是脂肪酸β氧化过程中的限速酶,而PGC-1α在预防NAFLD发病过程中起到重要作用,因此,两者的下调会促进肝脏的脂肪积累,导致肥胖和NAFLD的发生[12]。本研究还发现,SREBF-1的表达在IUGR子代中显著增加,这与Choi等[13]的研究结果一致。SREBF-2和HMGR、LDLR在肝脏胆固醇代谢中发挥重要作用。IUGR子代肝脏中,这3个基因均出现下调,这可能是由于胆固醇浓度增加引起的反馈性调节,降低胆固醇的生成,并减少从外周循环中摄入胆固醇[14]。
Rueda-Clausen等[3]的研究显示,高脂饮食诱导下,正常和IUGR大鼠均出现血清TG水平升高。然而,另一项研究并未发现这种现象,且在高脂饮食下的正常雄性大鼠中,还出现了血清TG水平的下降[4]。本研究中,在高脂饮食下,正常和IUGR大鼠均出现了血清TG水平的下降。而与血清TG水平相反的,肝脏组织中出现了脂肪堆积,且在IUGR子代中更为显著。这可能与肝脏组装和分泌极低密度脂蛋白的功能障碍有关。此外,SREBF-1在高脂饮食下表达增加,因此,肝内TG合成增加和分泌减少的联合作用可能是脂肪在肝脏中堆积以及外周血TG水平下降的重要原因。
在高脂饮食条件下,正常和IUGR大鼠均出现PPARα、ABCG5和CYP7A1的上调。在肝脏组织内,PPARα表达增加能够缓解NAFLD的进展,而ABCG5和CYP7A1分别在胆固醇分泌和转化成胆汁酸的过程中起到关键作用[15]。这些基因在转录水平的表达改变说明机体对不利饮食因素呈现出一定的抵抗作用,这也与既往在NAFLD病人中发现ABCG5表达显著升高的研究结果相似[16]。本研究还发现,IUGR和高脂饮食对LDLR存在交互作用,这可能是正常和IUGR大鼠应对高脂饮食的不同代偿机制造成的。
综上,本研究发现了IUGR大鼠在不同饮食环境中呈现不同的生长趋势,但均表现出糖脂代谢异常。这些异常表现可能与IUGR大鼠的食欲增强和转录水平上肝脏脂肪酸氧化功能障碍有关。高脂饮食条件下,血清TG下降而肝脏脂肪堆积,这可能与转录水平上肝脏脂肪酸合成增加和TG分泌减少相关。在幼年大鼠,对于IUGR和高脂饮食的不利影响,机体通过相关基因的转录调节呈现一定的抵抗作用,但并不足以逆转代谢紊乱。
| [1] | 儿童代谢综合征中国工作组. 中国六城市学龄儿童代谢综合征流行现状研究[J]. 中华儿科杂志, 2013, 51(6): 409-413. |
| [2] | Salam RA, Das JK, Bhutta ZA. Impact of intrauterine growth restriction on long-term health[J]. Curr Opin Clin Nutr Metab Care, 2014, 17(3): 249-254. |
| [3] | Rueda-Clausen CF, Dolinsky VW, Morton JS, et al. Hypoxia-induced intrauterine growth restriction increases the susceptibility of rats to high-fat diet-induced metabolic syndrome[J]. Diabetes, 2011, 60(2): 507-516. |
| [4] | Zinkhan EK, Chin JR, Zalla JM, et al. Combination of intrauterine growth restriction and a high-fat diet impairs cholesterol elimination in rats[J]. Pediatr Res, 2014, 76(5): 432-440. |
| [5] | Enjoji M, Yasutake K, Kohjima M, et al. Nutrition and nonalcoholic fatty liver disease: the significance of cholesterol[J]. Int J Hepatol, 2012, 2012: 925807. |
| [6] | Neitzke U, Harder T, Plagemann A. Intrauterine growth restriction and developmental programming of the metabolic syndrome: a critical appraisal[J]. Microcirculation, 2011, 18(4): 304-311. |
| [7] | 郑锐丹, 汪无尽, 应艳琴, 等. 生长追赶宫内发育迟缓大鼠早期糖脂代谢及脂肪细胞功能的改变[J]. 中国当代儿科杂志, 2012, 14(7): 543-547. |
| [8] | Remacle C, Bieswal F, Bol V, et al. Developmental programming of adult obesity and cardiovascular disease in rodents by maternal nutrition imbalance[J]. Am J Clin Nutr, 2011, 94(6 Suppl): 1846S-1852S. |
| [9] | Lim JS, Lee JA, Hwang JS, et al. Non-catch-up growth in intrauterine growth-retarded rats showed glucose intolerance and increased expression of PDX-1 mRNA[J]. Pediatr Int, 2011, 53(2): 181-186. |
| [10] | Fukami T, Sun X, Li T, et al. Mechanism of programmed obesity in intrauterine fetal growth restricted offspring: paradoxically enhanced appetite stimulation in fed and fasting states[J]. Reprod Sci, 2012, 19(4): 423-430. |
| [11] | Coupe B, Grit I, Hulin P, et al. Postnatal growth after intrauterine growth restriction alters central leptin signal and energy homeostasis[J]. PLoS One, 2012, 7(1): e30616. |
| [12] | Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease[J]. Int J Mol Sci, 2014, 15(5): 8713-8742. |
| [13] | Choi GY, Tosh DN, Garg A, et al. Gender-specific programmed hepatic lipid dysregulation in intrauterine growth-restricted offspring[J]. Am J Obstet Gynecol, 2007, 196(5): e471-e477. |
| [14] | Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis[J]. Prog Lipid Res, 2013, 52(1): 175-191. |
| [15] | Giby VG, Ajith TA. Role of adipokines and peroxisome proliferator-activated receptors in nonalcoholic fatty liver disease[J]. World J Hepatol, 2014, 6(8): 570-579. |
| [16] | Nakamuta M, Fujino T, Yada R, et al. Impact of cholesterol metabolism and the LXRalpha-SREBP-1c pathway on nonalcoholic fatty liver disease[J]. Int J Mol Med, 2009, 23(5): 603-608. |