 2015, Vol. 17
2015, Vol. 17



随着对儿童白血病机制研究的不断深入、危险度分组不断细化以及化疗、特异靶向治疗、干细胞移植等技术的不断提高,近年来儿童急性淋巴细胞性白血病(acute lymphoblastic leukemia,ALL)的疗效不断提高,5 年无事件生存率(EFS)达到80%以上[1]。近10 年来,通过不同手段监测患儿不同阶段的微小残留病(MRD)变化越来越成为指导患儿危险度划分及治疗的关键因素。随着基因检测手段的不断进步,IKZF1 基因在ALL发生发展中的地位日益突显。
人类IKZF1(ikaros family zinc finger 1)基因定位于7p12,DNA 序列全长125 kb,共8 个外显子,其编码蛋白IKAROS 是一种极为重要的造血转录调控因子,属锌指蛋白家族成员,尤其在淋巴系造血方面发挥着关键性调控作用[2, 3, 4]。IKZF1基因的缺失曾在鼠的T-ALL细胞株上被检测出来,在人类,该基因的缺失或突变多发生于B 系ALL(B-ALL),尤其是BCR/ABL 阳性ALL[5, 6, 7]。有文献报道:无论是儿童还是成人,BCR/ABL 阳性ALL 患者IKZF1 基因缺失都是最常见的遗传学改变(发生率高达83.7%,在成人和儿童患者中分别为90.9% 和76.2%)[8]。那么,对于BCR/ABL 阴性B-ALL 患儿,其IKZF1 基因的拷贝数情况是怎样的?该基因的拷贝数异常是否会对该部分患儿的预后产生影响?关于此部分内容国内未见报道。
为了解IKZF1 基因拷贝数在BCR/ABL 阴性B-ALL 患儿中的异常情况及该变化对此部分患儿预后的影响,本研究应用多重连接探针扩增(MLPA)技术对2008 年4 月至2013 年4 月我中心接受CCLG-ALL2008 方案治疗的180 例BCR/ABL 阴性B-ALL 患儿初诊时的DNA 标本进行检测,并回顾性分析该部分患儿的临床特点及预后,加深对我国BCR/ABL 阴性B-ALL 患儿发病特点的了解,寻找更加敏感实用的MRD 监测指标及更加经济有效的MRD 监测手段,进一步提高ALL 患儿的疗效。
1 资料与方法 1.1 研究对象我院2008 年4 月至2013 年4 月的初诊并可获得足够骨髓DNA 量的B-ALL 患儿共214 例,其中BCR/ABL 阴性B-ALL 患儿180 例(男112 例,女68 例),中位年龄4.0(1.0~15.0)岁。所有患儿均满足CCLG-ALL2008 方案纳入标准[9]。同时选取20 例健康志愿儿童作为正常对照,其中男10 例,女10 例,中位年龄7.0(3.1~14.5)岁。纳入本研究的患儿及正常儿童志愿者的监护人均知情同意。
1.2 诊断及分型标准参照1987 年全国白血病化疗讨论会制定的白血病诊断标准[10]。危险度分组标准参照CCLGALL2008方案危险度分组标准[11, 12]。MRD 分组标准[13]:应用流式细胞检测技术检测患儿诱导治疗后(d33)和巩固治疗期间(d88)骨髓肿瘤细胞残留情况。MRD 标危组(MRD-SR):d33 和d88 MRD 均为阴性;MRD 高危组(MRD-HR):d33 MRD 水平≥ 10-2 或d88 MRD ≥ 10-3;MRD 中危组(MRD-IR):不符合标危组和高危组者入中危组。脑脊液状态分级主要依据临床表现、影像学改变、脑脊液细胞流式细胞检测技术或目力计数及脑脊液形态学(离心涂片法),分为CNS1 状态(无中枢神经系统白血病)、CNS2 状态(脑脊液可见到明确的白血病细胞或初次腰穿脑脊液混血)及CNS3 状态(中枢神经系统白血病)。
1.3 MLPA 检测初诊时留取患儿单个核细胞,标本收集达一定数量后统一提取DNA 并行MLPA 检测。
单个核细胞提取:取肝素抗凝骨髓3~5 mL,经PBS 稀释后,用Ficoll 淋巴细胞分离液分离单个核细胞,1 100~1 200 转/min,离心11~12 min。吸取中间细胞层,用磷酸盐缓冲液(PBS)洗涤2 次,800~1 000 转/min,离心7~8 min,弃上清。计数细胞,分装,冻存 -20℃冰箱备用。按试剂盒步骤提取DNA。按照SALSA MLPA KIT P335-B1 ALL-IKZF1试剂盒说明书进行IKZF1 各外显子拷贝数缺失情况的检测。
(1)DNA 变性:取5 μL(20 ng/μL)的样品,98℃处理5 min,25℃暂停。
(2) 探针与DNA 的杂交: 取出样品,降至室温,加入3 μL 的杂交混合液(1.5 μL SALSAprobemix+1.5 μL MLPA 缓冲液),95 ℃ 1 min,60℃温浴16~24 h。
(3)杂交探针的连接:PCR 仪降到54℃,打开管盖; 加入32 μL 的连接酶混合液(3 μLligase buffer A+3 μL ligase buffer B+25 μL 蒸馏水 +1 μL ligase-65(绿盖)。54℃温浴15 min;98℃加热5 min 灭活连接酶后20℃暂停。
(4)连接探针的PCR 扩增:从PCR 仪中取出样品,降至室温;加入10 μL PCR 混合液(7.5 μL蒸馏水+2 μL PCR Primer Mix +0.5 μL SALSAPolymerse);PCR 扩增:95 ℃,30 s → 60 ℃,30 s → 72 ℃,60 s,35 个循环后,72 ℃,20 min → 15℃暂停。
(5)PCR 产物的毛细管电泳:0.5 μL 样品+0.5 μL 500LIZ® Size Standard+9 μL Hi-DiTMFormamide。所得结果通过GeneMapper 4.0 软件和Coffalyser 软件进行半定量分析[13]。
根据有无IKZF1 缺失将180 例患儿分成两组:“IKZF1 缺失组”和“IKZF1 正常组”。IKZF1 基因8 个外显子中任意1 个或多个外显子发生缺失者进入“IKZF1 缺失组”;而IKZF1 基因无任何拷贝数异常者进入“IKZF1 正常组”。
1.4 化疗方案及疗效评估参照CCLG-ALL2008 化疗方案[11, 12] 进行化疗。疗效评估参照1987 年全国白血病化疗讨论会制定的白血病治疗疗效标准[14]。无病生存(diseasefreesurvival,DFS)期指患者从完全缓解(CR)开始至复发或缓解期死亡的时间。总生存(overallsurvival,OS)期指自诊断到死亡或末次随访日期。随访时间截至2015 年4 月30 日,失访者截止至失访日期。中位随访时间38 个月(范围1~84 个月)。
1.5 统计学分析应用SPSS 17.0 统计软件进行统计学处理与分析。计量资料采用均值± 标准差(x±s)表示,两组间比较采用成组t 检验;计数资料采用百分率(%)表示,组间比较采用卡方检验。生存曲线采用Kaplan-Meier 分析,多因素分析采用Cox 回归分析。P<0.05 为差异有统计学意义。
2 结果2.1 MLPA 检测结果
对正常对照样本的结果进行分析,对照样本实验的重复性较好,且比较稳定,每一个样本的内标均已达到检测结果可信度高的范围,通过箱式图可以直观明了地对IKZF1 缺失或扩增进行判断。180 例患儿中,27 例患儿发生了IKZF1 缺失,缺失率为15.0%。其中IKZF1 基因8 个外显子全部缺失者4 例,单纯1 号外显子缺失者14 例,4~7 号外显子缺失者3 例,2~7 号外显子缺失者2 例,3~7 号外显子缺失者1 例,1、8 号外显子缺失者3 例。未检出基因扩增现象。
2.2 两组患儿临床特征的比较IKZF1 缺失组患儿初诊时白细胞水平及流式MRD- 高危组患儿的比例明显高于IKZF1 正常组;IKZF1 缺失组患儿多发生在无特殊融合基因异常的BCR/ABL 阴性患儿,且IKZF1 基因缺失患儿易伴随出现11、8、5、7、21 号等染色体的异常。而两组患儿在性别、年龄、初诊时脑脊液状态、泼尼松试验反应情况以及根据CCLG-2008 方案划分的最终危险度分组比例均无明显差异(表 1)。
|
|
表 1 IKZF1 缺失组与IKZF1 正常组之间临床特征比较 |
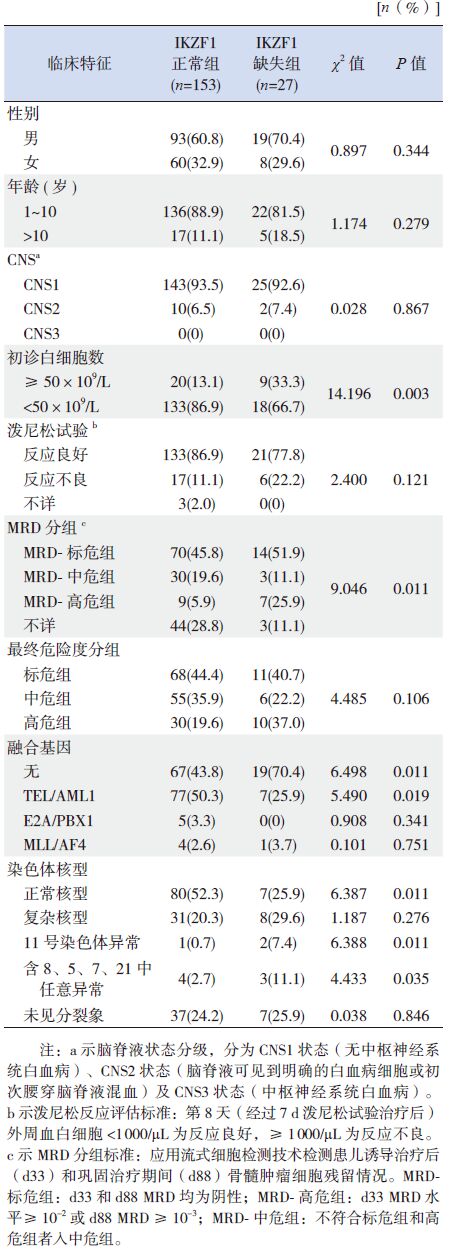
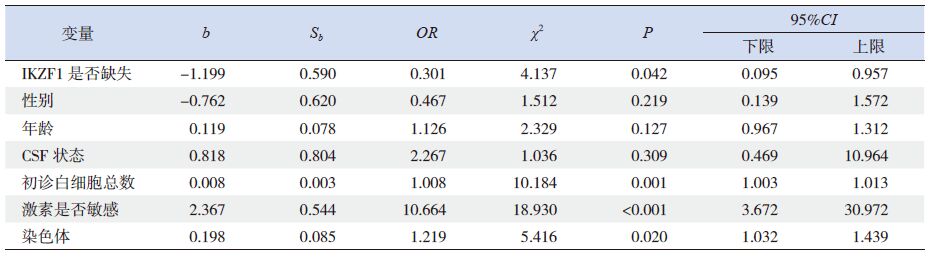
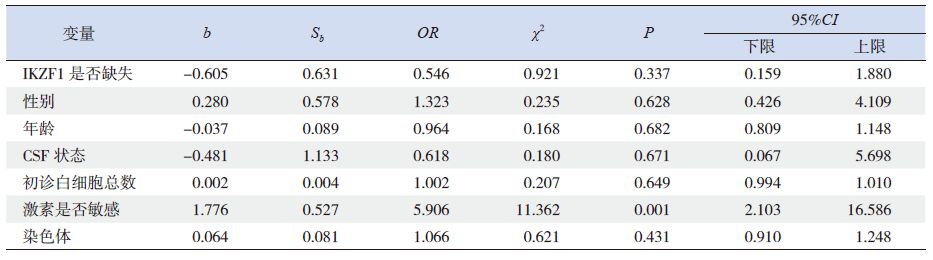
应用Kaplan-Meier 法分析IKZF1 缺失与否对180 例BCR/ABL 阴性B-ALL 患儿DFS 及OS 的影响。结果显示:IKZF1 缺失组其DFS 率明显低于IKZF1正常组(0.740±0.096 vs 0.905±0.034,P=0.002)(图 1);而两组患儿OS 率差异无统计学意义(0.849±0.070 vs 0.930±0.021,P=0.148)(图 2)。应用Cox 法分析IKZF1 缺失与否对BCR/ABL 阴性B-ALL 患儿DFS 及OS 的影响,结果显示:IKZF1缺失不利于患儿的DFS(P<0.05),但对患儿的OS 无明显影响(表 2、3)。比较两组复发患儿情况发现,IKZF1 缺失组患儿中5 例复发,复发率18.5%(5/27),均为骨髓复发;而IKZF1 正常组共有8 例患儿复发,复发率5.2%(8/153),其中4 例(50%)为骨髓复发,4 例(50%)为髓外复发(其中中枢神经系统复发2 例,睾丸复发2 例)。

|
图 1 IKZF1 缺失组与IKZF1 正常组患儿3 年DFS 比较 |

|
图 2 IKZF1 缺失组与IKZF1 正常组患儿3 年OS 比较 |
|
|
表 2 Cox 法分析IKZF1 缺失与否对BCR/ABL 阴性B-ALL 患儿DFS 的影响 |
|
|
表 3 Cox 法分析IKZF1 缺失与否对BCR/ABL 阴性B-ALL 患儿OS 的影响 |
IKZF1 基因编码IKaros 蛋白。Ikaros 蛋白定位于细胞核,是多能干细胞发育为成熟淋巴细胞的关键。体外实验显示,Ikaros 蛋白可以诱导B、T 淋巴细胞分化,调控细胞凋亡及细胞周期。近期研究显示,大约10%~30% 的B-ALL 有IKZF1 基因的失活,其中多数为IKZF1 基因的缺失,小部分为突变(无意义、错义或导致无活性的Ikaros蛋白)[6]。且该基因的缺失多与BCR/ABL 阳性伴随发生[15]。van der Veer 等[16] 报道IKZF1 基因的状态对于BCR/ABL 阳性B-ALL 患儿的预后具有指导意义。那么,IKZF1 基因的改变在BCR/ABL 阴性B-ALL 患儿中的表达情况是怎样的?它对于该部分患儿的预后是否存在一定的价值呢?国内目前未见报道。
本研究对我院初诊并可获得足够骨髓DNA 量的B-ALL 患儿214 例进行IKZF1 基因的检测,其中BCR/ABL 阴性B-ALL 患儿180 例。本研究显示180 例患儿中共有27 例发生了IKZF1 缺失,缺失率为15.0%,与国际报道的IKZF1 基因在B-ALL中的缺失率(10%~30%)相一致[6]。根据是否存在IKZF1 缺失将180 例患儿分为IKZF1 缺失组和IKZF1 正常组后比较两组临床资料发现,IKZF1 缺失组患儿初诊时白细胞水平及流式MRD- 高危组患儿的比例明显高于IKZF1 正常组。该结果间接提示IKZF1 缺失患儿肿瘤细胞增殖速度明显高于IKZF1 正常组。同时本研究发现IKZF1 基因缺失患儿易伴随出现11、8、5、7、21 号等染色体的异常。该结果提示该IKZF1 基因缺失与附加染色体异常相伴随,具体机制尚需进一步研究。且上述染色体异常多发生于骨髓增生异常综合征(myelodysplasticsyndrome,MDS)患者中,是否提示IKZF1 基因缺失患儿多伴有MDS 经过?有待进一步研究证实。应用Kaplan-Meier 法分析IKZF1 缺失与否对180例BCR/ABL 阴性B-ALL 患儿DFS 及OS 的影响,结果显示IKZF1 缺失组DFS 明显低于IKZF1 正常组,Cox 法多因素回归分析同样支持IKZF1 缺失对BCR/ABL 阴性B-ALL 的DFS 存在独立的影响。该结果提示,IKZF1 基因缺失与否对于患儿的缓解期长短有重要的预示作用,是独立于目前已知危险因素以外的重要指标。但Vitanza 等[17] 报道单纯敲除IKZF1 基因并不能导致BCR/ABL 阴性B-ALL 患儿发生耐药,提示IKZF1 基因缺失可能与其他基因协同发生作用。在本研究中,IKZF1 基因缺失与否虽然与患儿的DFS 密切相关,但本研究未发现IKZF1 基因缺失对患儿的长期生存产生统计学意义的影响,可能与病例数量有限、随访时间不够长、医疗水平提高以及人民生活水平提高等因素导致复发患儿存活率提高有关。本研究还显示,IKZF1 缺失组患儿均为骨髓复发,而IKZF1 正常组有50% 为髓外复发(其中中枢神经系统复发2 例,睾丸复发2 例),提示IKZF1 缺失可能对于患儿骨髓肿瘤干细胞有更加重要的影响。
综上所述,BCR/ABL 阴性B-ALL 患儿存在一定比例的IKZF1 缺失,该基因缺失往往提示患儿具有较短的缓解期。适当加大该部分患儿的化疗剂量、缩短化疗间期可能有望延长这部分患儿的缓解时间,为其争取更多的治疗机会,得到更加满意的治疗效果。MLPA 方法检测基因拷贝数变化,方法简单快捷,成本适中,为ALL 患儿MRD 的监测提供了新的可能检测手段。
| [1] | Ribera J, Morgades M, Zamora L, et al. Prognostic significance of copy number alterations in adolescent and adult patients with precursor B acute lymphoblastic leukemia enrolled in PETHEMA protocols[J]. Cancer, 2015, 121(21): 3809-3817. |
| [2] | Olsson L, Johansson B. Ikaros and leukaemia[J]. Br J Haematol, 2015, 169(4): 479-491. |
| [3] | Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment[J]. Science, 1992, 258(5083): 808-812. |
| [4] | Medina KL, Pongubala JM, Reddy KL, et al. Assembling a gene regulatory network for specification of the B cell fate[J]. Dev Cell, 2004, 7(4): 607-617. |
| [5] | Iacobucci I, Storlazzi CT, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell'Adulto Acute Leukemia Working Party (GIMEMA AL WP)[J]. Blood, 2009, 114(10): 2159-2167. |
| [6] | Medeiros BC. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia[J]. N Engl J Med, 2009, 360(17): 1787. |
| [7] | Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study[J]. Lancet Oncol, 2009, 10(2): 125-134. |
| [8] | Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros[J]. Nature, 2008, 453(7191): 110-114. |
| [9] | Ofverholm I, Tran AN, Heyman M, et al. Impact of IKZF1 deletions and PAX5 amplifications in pediatric B-cell precursor ALL treated according to NOPHO protocols[J]. Leukemia, 2013, 27(9): 1936-1939. |
| [10] | 张之南, 沈悌. 血液病诊断及疗效标准[M]. 第3版. 北京: 科学出版社, 2007: 19-23. |
| [11] | Gao C, Zhao XX, Li WJ, et a1. Clinical features, early treatment responses, and outcomes of pediatric acute lymphoblastic leukemia in China with or without specific fusion transcripts: a single institutional study of 1,004 patients[J]. Am J Hematol, 2012, 87(11): 1022-7. |
| [12] | 刘晓明, 邹尧, 王慧君, 等. CCLG-2008方案治疗标危中危儿童急性淋巴细胞白血病中期随访结[J]. 中华儿科杂志, 2014, 52(6): 449-454. |
| [13] | Dörge P, Meissner B, Zimmermann M, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol[J]. Haematologica, 2013, 98(3): 428-432. |
| [14] | 张之南, 沈悌. 血液病诊断及疗效标准[M]. 第3版. 北京: 科学出版社, 2007: 131-132. |
| [15] | Martinelli G, Iacobucci I, Storlazzi CT, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report[J]. J Clin Oncol, 2009, 27(31): 5202-5207. |
| [16] | van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL[J]. Blood, 2014, 123(11): 1691-1698. |
| [17] | Vitanza NA, Zaky W, Blum R, et al. Ikaros deletions in BCR-ABL-negative childhood acute lymphoblastic leukemia are associated with a distinct gene expression signature but do not result in intrinsic chemoresistance[J]. Pediatr Blood Cancer, 2014, 61(10): 1779-1785. |