 2015, Vol. 17
2015, Vol. 17



2. 上海交通大学医学院附属儿童医院肾脏风湿科, 上海 200040
Dent病(Dent disease, OMIM 300009)是一种罕见的X连锁隐性遗传性肾小管疾病,其主要临床特点包括低分子量蛋白尿(low-molecular-weight proteinuria, LMWP)、高钙尿症、肾脏钙质沉着或肾结石等,部分患者可出现进行性肾功能不全,甚至肾衰竭。近年来,疾病靶向序列测序等基因诊断手段的迅速发展,有利于罕见病例的诊断。佝偻病是Dent病的常见并发症[1],但各国报道的佝偻病并发率不一。欧美Dent病患者佝偻病并发率为33%左右[2],而日本只有2%[3]。我国Dent病患者佝偻病并发率尚不详,目前尚无表现为Fanconi综合征的Dent病例的报道。我院收治的4例Dent病患儿中3例(75%)并发佝偻病,且3例表现为Fanconi综合征,现将这4例Dent病患儿的临床及基因资料总结并报告如下。
1 资料与方法 1.1 研究对象中国医学科学院北京协和医院2014年2~10月收治的Dent病患儿4例纳入研究。4例患儿来自4个不同的家庭。所有患儿均符合以下3条临床诊断标准[1, 4]:(1)低分子量蛋白尿明显增高:尿β2微球蛋白、α1微球蛋白和(或)视黄醇结合蛋白(RBP)等低分子尿蛋白增高达正常上限的5倍以上;(2)高钙尿症:24 h尿钙>4 mg/kg或尿钙/肌酐比>0.25 mg/mg;
(3)下列情况至少满足1项:肾钙质沉着、肾石症、血尿、低磷血症、肾功能不全。
1.2 基因检测经患儿及其监护人知情同意后,分别采集静脉血5 mL,并行CLCN5和OCRL1基因检测。基因检测由独立的基因检验所完成。4例患儿的CLCN5基因分析引物序列如表 1。
| 表 1 4 例Dent 病患儿的CLCN5 基因分析引物序列 |

4例患儿的主要临床资料和实验室检查结果见表 2。4例患儿均为男孩。就诊年龄为生后3 d至7岁,确诊年龄为4.5~12岁。就诊到确诊的间隔时间4.5~7.8年不等,平均6.1±1.6年。其中例1~3的首发症状均为多饮、多尿、双下肢呈X型,例3 存在轻度智力障碍和白内障。例1~3外院均曾诊断为“Fanconi综合征”,并予磷酸盐合剂及对症治疗,效果不佳。例4 在生后3 d时偶然查尿常规发现蛋白尿和血尿,之后监测蛋白尿和血尿持续存在。2岁时,其尿蛋白达到1 g/d,肾脏B超提示肾钙质沉着,肾脏病理提示为局灶性节段性肾小球硬化。诊断为“肾病综合征”,给予环孢素A(cyclosporine A, CsA)和泼尼松治疗1年余,蛋白尿曾达到部分缓解。但复查发现血浆肌酐从20 μmol/L进行性升高至72 μmol/L,因而将CsA减量。当CsA剂量下降后,血肌酐水平恢复正常,但患儿再次出现大量蛋白尿而至我院就诊。
| 表 2 4 例Dent 病患儿主要临床特点和实验室检查 |
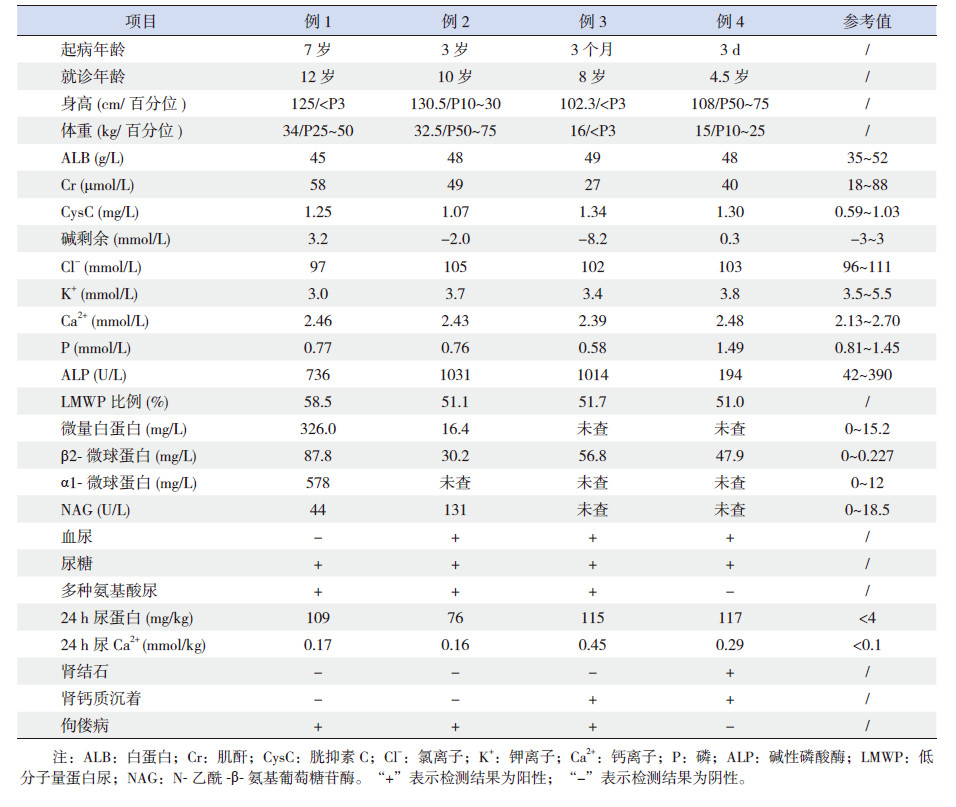
4例患儿均有明显增高的低分子量蛋白尿和高钙尿症。其中3例有血尿,多为镜下血尿,例4曾有一过性肉眼血尿,肾结石1例,肾钙质沉着2例,低磷血症3例,佝偻病3例。
例2母孕期羊水增多,余无特殊。例1和例3都曾有多次外伤后骨折史,余无特殊。
2.2 基因检测结果基因检测结果显示,例1存在CLCN5基因的缺失突变exon 6-7del,其父母均未行基因检测。例2存在CLCN5基因的缺失突变c.785_787del(p.263del Leu),其父母在该位点均未发现相同突变,为新生突变(图 1)。例3 未发现CLCN5和OCRL1等基因的致病突变。例4在CLCN5基因上发现一个无义突变c.1039 C>T(p.Arg347Term),其父母在该位点未发现相同突变,为新生突变(图 2)。c.1039 C>T(p.Arg347Term)突变曾有报道[5],而exon 6-7del和c.785_787del(p.263del Leu)是未报道过的,为新突变。
 |
图 1 例2 患儿及家系CLCN5 基因的部分一代测序 图谱 A: 患儿在CLCN5 基因上存在缺失突变c.785_787del (p.263del Leu),箭头所指为突变位点;B:患儿父亲在该位点 未发现突变;C:患儿母亲基因型为野生型。 |
 |
图 2 例4 患儿及家系CLCN5 基因的部分一代测 序图谱 A:患儿在CLCN5 基因上存在无义突变c.1039 C>T (p.Arg347Term),箭头所指为突变位点;B:患儿父亲在该位点 未发现突变;C:患儿母亲基因型为野生型。 |
目前Dent病的发病率尚不清。至2014年,全球报道了300多个Dent 病家系[6]。约60% 的Dent病患者的突变基因在CLCN5基因,15%的突变基因在OCRL1基因,剩余的25%的患者尚未发现致病基因[1]。人类基因突变数据库到2015年1月为止报道的1型Dent病相关的CLCN5突变有183种。Mansour-Hendili等[6]报道到2014年10月为止,检索PubMed数据库发现192种CLCN5基因突变,其中大缺失占4%,无义突变占17%,移码突变导致编码提前终止占28%,剪切突变占11%,错义突变占36.5%。人类基因突变数据库到2015年1月为止报道的2型Dent病相关的OCRL1突变有29种左右。目前尚未发现热点突变。基因型和临床表型的相关性还不确定。本研究中Dent病患儿的CLCN5基因突变分别为exon 6-7del,c.785_787del和c.1039 C>T。其中,exon 6-7del和c.785_787del是本研究报道的新突变。exon 6-7del为外显子6~7的纯合缺失,这样大片段的缺失可使CLC-5蛋白缺失功能或导致蛋白质产物降解;c.785_787del突变导致第263位氨基酸缺失,可导致蛋白质空间构象变化,从而导致蛋白质功能缺陷;c.1039 C>T为纯合的无义突变,位于外显子8,导致基因编码的氨基酸在第347位提前终止,可引起蛋白质产物降解。
目前Dent病的发病机制尚无定论。目前认为,CLCN5基因位于染色体Xp11.23-p11.22,有12个外显子,编码746个氨基酸长度的CLC-5蛋白。该蛋白为一种2Cl-/H+交换体[7, 8],可在肾脏近端肾小管、皮质集合小管和髓襻升支粗段表达。正常情况下,原尿中的低分子尿蛋白在近端小管上皮经Megalin/Cubilin等受体介导的胞吞作用而被重吸收。近端小管上皮细胞的内涵体(endosome)中pH和(或)Cl-浓度在这一过程中起关键作用。而CLC-5蛋白与H-ATPase等共同调控内涵体的pH及Cl-浓度。一旦CLC-5蛋白的结构异常,低分子蛋白尿的重吸收受阻,低分子量尿蛋白则明显增高。此外,甲状旁腺素(PTH)重吸收受阻,尿中高浓度的PTH可刺激VitD3合成从而促进肠道吸收钙增加,激活肾小管顶端膜PTH受体活化刷状缘的钠-磷共转运蛋白等途径导致高尿钙及高磷酸盐尿等。
OCRL1基因位于染色体Xq24-26,有24个外显子。编码生成的OCRL1蛋白具有磷酸脂酶活性,可以催化水解磷脂酰肌醇4,5-二磷酸(PIP2)等[1]。OCRL1蛋白分布在肾小管上皮细胞的溶酶体、内涵体和高尔基复合体。具体致病机制尚不明确。OCRL1蛋白在全身多处均有分布,除了肾脏外,在眼晶状体和脑组织等均有分布。因此,2型Dent病除肾脏表现外,还可能出现轻度智力发育落后和白内障等肾外表现。
国外报道的Dent病的蛋白尿多为轻中量,但本研究中4例患儿均为大量蛋白尿。经检索PubMed和万方、CNKI数据库,到2015年6月1日为止,我国共发表了4篇9例Dent病,其中1型Dent病8例,2型Dent病1例[9, 10, 11, 12]。9例中有4例为大量蛋白尿,3例为中量蛋白尿。蛋白尿程度不同考虑与肾脏受累程度不同有关。Dent病初期以肾小管性蛋白尿为主,但随着病程发展,均可出现肾小球损害而出现大量蛋白尿。但Dent病是以肾小管性蛋白尿为主,因此即使有大量蛋白尿,一般白蛋白不低,也没有水肿等表现。2013年Edvardsson等[13]推荐了 Dent病的诊断流程图,其中低分子蛋白尿增高是诊断Dent病的关键线索。因此对初诊蛋白尿的患儿,尤其是血浆白蛋白没有明显下降的,建议完善尿蛋白电泳等检查明确尿蛋白成分及性质,必要时进一步基因诊断以协助确诊。
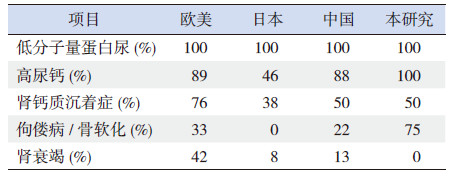
包括Edvardsson等[13]推荐的 Dent病的诊断流程都以蛋白尿为切入点。但需注意的是,单纯的肾小管性蛋白尿的临床症状往往并不明显。所以类似诊断流程更适合日韩等已建立学龄儿童等的常规尿检体系的国家或城市,或者偶然查尿常规发现蛋白尿的患儿,如本组病例中的例4。为了提高对我国1型Dent病的患儿的认识,本研究比较了欧美国家、日本、我国目前报道文献及本组数据(表 3)[2, 3],发现我国Dent病患儿的主要临床表现与国外文献报道一致,均为低分子量蛋白尿、高尿钙、肾钙质沉着等。但有意思的是,佝偻病是Dent病的常见并发症,但各国报道的发生率差异较大。欧美的Dent患者有1/3左右并发佝偻病,日本的Dent病患儿佝偻病的发生率则非常低,而中国已报道的Dent病患儿佝偻病的发生率介于两者之间,而本研究患儿佝偻病的发生率达75%,是其中最高的,且其中有3例表现为Fanconi综合征。而日本的Dent病患儿几乎没有表现为Fanconi综合征的,国内之前也没有表现为Fanconi综合征的Dent病患儿的报道。分析其原因,考虑除了种族、环境、饮食习惯等因素外,还与对患儿的发现途径不同有关。日韩等国的Dent病患儿多因尿检筛查异常而发现,可以做到早期发现、早期干预[3]。我们推测早期干预减少了日本等国Dent病患儿Fanconi综合征等较严重肾小管损害表现的发生率。本组4例Dent病患儿中有3例是从首发表现为Fanconi综合征的人群中发现的。儿童Fanconi综合征大多与遗传有关。Dent病等原发性近端肾小管功能受累引起的Fanconi综合征表现通常发生在病程早期 [14]。因此,本研究提示了Dent病诊断的一种新思路:对于同时存在高尿钙、肾脏钙质沉着或肾结石等Fanconi综合征患儿,应该考虑到Dent病的可能性。与典型的Fanconi综合征患儿有所不同的是,本组Dent病患儿较少出现代谢性酸中毒,且没有高氯血症。
| 表 3 欧美、日本、中国和本研究中1 型Dent 病患儿的临床特点 |
例4在疾病初期曾被诊断为肾病综合征,给予CsA和泼尼松治疗。朱碧溱等[12]也报道,Dent病患儿在疾病初期大多被误诊为肾病综合征或肾炎,给予多种免疫抑制剂治疗无效。免疫抑制剂的使用不仅增加了家庭的经济负担,更重要的是可能导致感染、生长发育落后等多种严重不良反应。因此,早期诊断以避免不必要的治疗对Dent病患儿非常重要。
关于Dent病的肾脏病理方面,目前资料尚有限。目前国内报道的9例Dent病患儿中有3例做了肾活检,提示肾小管间质轻度损伤1例,局灶性肾小球硬化1例,轻微病变1例。本组病例中例4的肾脏病理提示为局灶性节段性肾小球硬化(FSGS)。国外文献报道,Dent病的肾脏病理在光镜下多为非特异性病变,肾小管间质损害从无明显变化到显著的纤维化都可能存在,部分肾脏髓质有钙化,多有单核细胞浸润,免疫复合物或补体沉积一般为阴性;电镜下近端小管细胞的超微结构多没有明显异常[1]。 Dent病是FSGS的病因之一[1, 15, 16]。Dent病患儿肾脏病理表现为FSGS时,临床上多表现为肾病水平的蛋白尿。
治疗方面,Dent病目前尚无分子靶向治疗,无法治愈。治疗上以支持治疗为主,重点在于降低尿钙水平,防治肾结石,延缓肾功能衰竭的发生和进展。可通过多饮水、噻嗪类利尿剂等降尿钙。小剂量的氢氯噻嗪(每天0.4~0.8 mg/kg)可以降低尿钙的排泄,但可出现脱水、低钾血症、低钠血症等不良反应,所以儿童须谨慎使用[17]。对CLC-5基因敲除小鼠研究发现,长期高枸橼酸饮食可减少尿钙排泄,从而减少肾钙化沉着,延缓肾功能不全的发展[1]。合并佝偻病表现的患儿应加用磷酸盐合剂和活性VitD3的治疗。VitD3治疗期间尤其须注意监测尿钙,因其可促进钙吸收从而可能增加尿钙的排泄,从而加重肾损害。文献报道,肾移植术后的Dent病患儿的移植肾未再复发肾结石、肾钙质沉积症[2]。因此肾移植可以作为Dent病肾衰竭患儿的一种治疗选择。
本组病例中的例4在病程中曾用CsA治疗达到蛋白尿部分缓解,考虑可能机制与CsA治疗Alport综合征的相似。患儿蛋白尿的减少可能并不是基于CsA的免疫抑制作用,而是基于钙调神经磷酸酶抑制剂具有稳定足细胞骨架的非免疫作用[18]。由于CsA可能存在的肾毒性,能否用于Dent病的治疗,还需要更多的研究证据支持。
欧美国家约30%~80%的Dent病患者在20~40岁左右进入慢性肾功能不全[1],但日本等报道的肾衰竭发生率仅8%左右,这可能与日本Dent病患儿的早期发现和早期干预有关[3]。
目前75%左右的Dent病患儿可以在CLCN5或OCRL1基因上找到突变位点。但即使技术上可行,目前国外并不推荐Dent病行产前诊断和胚胎植入前的基因检测[1]。因为,大多数的Dent病患者的整个生命进程是好的,且目前的基因型和临床表型间尚未发现明确的相关性,同一家庭中的Dent病患者的临床表型可有显著的不同。
总之,对于蛋白尿患儿,不但要考虑肾小球性蛋白尿,还需考虑到肾小管性,应尽早完善尿蛋白电泳等检查明确尿蛋白的成分;对于Fanconi综合征患儿,如同时存在高尿钙、肾脏钙质沉着或肾结石等临床表现,应考虑到Dent病可能;诊断过程中,应以临床表征和病理为主导,行基因检测明确诊断,以实现合理的治疗。
| [1] | Devuyst O, Thakker RV. Dent's disease[J]. Orphanet J Rare Dis, 2010, 5:28-34. |
| [2] | Claverie-Martín F, Ramos-Trujillo E, García-Nieto V. Dent's disease:clinical features and molecular basis[J]. Pediatr Nephrol, 2011, 26(5):693-704. |
| [3] | Sekine T, Komoda F, Miura K, et al. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with lowmolecular-weight proteinuria[J]. Nephrol Dial Transplant, 2014, 29(2):376-384. |
| [4] | Hoopes RR, Raja KM, Koich A, et al. Evidence for genetic heterogeneity in Dent's disease[J]. Kidney Int, 2004, 65(5): 1615-1620. |
| [5] | Hoopes RR Jr, Shrimpton AE, Knohl SJ, et al. Dent disease with mutations in OCRL1[J]. Am J Hum Genet, 2005, 76(2):260-267. |
| [6] | Mansour-Hendili L, Blanchard A, Pottier NL, et al. Mutation update of the ClCN5 gene responsible for Dent disease 1[J]. Hum Mutat, 2015, 36(8):743-752. |
| [7] | Pusch M, Zifarelli G. ClC-5:Physiological role and biophysical mechanisms[J]. Cell Calcium, 2015, 58(1):57-66. |
| [8] | Novarino G, Weinert S, Rickheit G, et al. Endosomal chlorideproton exchange rather than chloride conductance is crucial for renal endocytosis[J]. Science, 2010, 328(5984):l398-1401. |
| [9] | Ji LN, Chen CY, Wang JJ, et al. A novel CLCN5 mutation in a Chinese boy with Dent's disease[J]. World J Pediatr, 2014, 10(3):275-277. |
| [10] | Zhang H, Wang C, Yue H, et al. Identification of a novel mutation in the CLCN5 gene in a Chinese family with Dent-1 disease[J]. Nephrology (Carlton), 2014, 19(2):80-83. |
| [11] | 李国民, 方晓燕, 徐虹, 等. 儿童2型Dent病1例并文献复习[J]. 中国循证儿科杂志, 2014, 9(6):456-459. |
| [12] | 朱碧溱, 李鹏, 黄建萍. 以小分子蛋白尿为主要表现的Dent病六例临床及基因分析[J]. 中华儿科杂志, 2010, 48(5):329-333. |
| [13] | Edvardsson VO, Goldfarb DS, Lieske JC, et al. Hereditary causes of kidney stones and chronic kidney disease[J]. Pediatr Nephrol, 2013, 28(10):1923-1942. |
| [14] | Solano A, Lew SQ, Ing TS. Dent-Wrong disease and other rare causes of the Fanconi syndrome[J]. Clin Kidney J, 2014, 7(4): 344-347. |
| [15] | Copelovitch L, Nash MA, Kaplan BS. Hypothesis:Dent disease is an underrecognized cause of focal glomerulosclerosis[J]. Clin J Am Soc Nephrol, 2007, 2(5):914-918. |
| [16] | Frishberg Y, Dinour D, Belostotsky R, et al. Dent's disease manifesting as focal glomerulosclerosis:is it the tip of the iceberg?[J]. Pediatr Nephrol, 2009, 24(12):2369-2373. |
| [17] | Blanchard A, Vargas-Poussou R, Peyrard S, et al. Effect of hydrochlorothiazide on urinary calcium excretion in Dent disease:an uncontrolled trial[J]. Am J Kidney Dis, 2008, 52(6): 1084-1095. |
| [18] | 张琰琴, 丁洁, 王芳, 等. Alport综合征治疗进展[J]. 中华儿科杂志, 2015, 53(1):76-77. |