 2015, Vol. 17
2015, Vol. 17



急性髓系白血病(acutemyeloidleukemia,AML)约占儿童急性白血病的15%~25%,是一组起源于造血干细胞的恶性克隆性疾病,具有高度异质性[1]。细胞遗传学分析有助于AML的分型,并且具有重要的预后价值[2]。儿童AML的细胞遗传学改变种类较多,常见并与儿童AML预后有关的有20余种[3]。由于性染色体缺失主要见于M2型,为探讨AML性染色体缺失与预后的关系,现对2002年4月至2013年12月本院儿科收治的初治AMLM2型患儿细胞遗传学表现与预后之间的关系进行分析,总结报告如下。 1 资料与方法 1.1 研究对象
选取2002年4月至2013年12月我院儿科收治的初治儿童AMLM2型120例为研究对象,其中男66例,女54例,中位年龄11岁(范围2~17岁)。所有患儿均经骨髓形态学、细胞组织化学染色、细胞免疫分型、细胞遗传学及分子生物学检测确诊,且所有患儿均完成了至少1个疗程化疗。120例患儿中,14例无细胞遗传学检测结果,余106例根据细胞遗传学结果分为:正常核型组(A组)、不伴性染色体缺失的非正常核型组(B组)和伴性染色体缺失的非正常核型组(C组)。 1.2 染色体制备及核型分析
取患者治疗前骨髓1~2mL,经短期培养法(24h)培养后收集有丝分裂期细胞,常规制片,R显带技术进行染色体显带,细胞核型异常依据《人类细胞遗传学国际命名体制(ISCN1995)》描述。 1.3 治疗方案
(1)诱导缓解治疗:采用双诱导方案:阿糖胞苷(Ara-C)[100~150mg/(m2·d)×7d]+柔红霉素(DNR)[40~60mg/(m2·d)×2d]或去甲氧柔红霉素(Idr)[10mg/(m2·d)×2d]+依托泊苷(VP16)[100~150mg/(m2·d)×3d],共2次。
(2)巩固强化治疗:采用以下3个方案序贯治疗12~18个月:①Ara-C[2g/(m2·d)×4d]+DNR[40~60mg/(m2·d)×2d]或Idr[10mg/(m2·d)×2d];②三尖杉酯碱[4~6mg/(m2·d)×7d]+Ara-C[100~150mg/(m2·d)×7d];③Ara-C[100~150mg/(m2·d)×7d]+DNR[40~60mg/(m2·d)×2d]或Idr[10mg/(m2·d)×2d]+VP16[100~150mg/(m2·d)×3d]。完成巩固化疗后停药随访,蒽环类药物累计总量约相当于DNR350mg/m2。
(3)中枢神经系统白血病的防治:鞘内注射Ara-C、地塞米松和甲氨蝶呤三联药物,在每次巩固化疗使用小剂量Ara-C[100~150mg/(m2·d)]时进行,总疗程内行鞘内注射4~8次。
(4)造血干细胞移植:23例患儿进行了造血干细胞移植。 1.4 疗效标准
疗效标准按照文献[4]标准进行评估。 1.5 随访
随访时间截至2013年12月31日。中位随访时间18个月(范围1~139个月)。无事件生存(EFS)期定义为从诊断到第1次事故(包括复发、在完全缓解期间的死亡)或末次随访的时间。总生存(OS)期指从诊断到各种原因死亡的时间。 1.6 统计学分析
采用SPSS20.0统计软件对数据进行统计学分析。不符合正态分布的计量资料采用中位数(范围)表示,多组间比较采用秩和检验;计数资料采用百分率(%)表示,多组间比较采用χ2检验;生存分析应用Kaplan-Meier生存曲线,多组生存率的比较应用log-rank检验。P<0.05为差异有统计学意义。 2 结果 2.1 一般情况
106例行细胞遗传学检测的患儿中,男60例,女46例;正常核型26例,发生率为24.5%;伴性染色体缺失的非正常核型28例,发生率为26.4%,其中Y染色体缺失17例,全部为男性,占总男性患儿的28%(17/60),X染色体缺失11例,全部为女性,占总女性患儿的24%(11/46);不伴性染色体缺失的非正常核型52例,发生率为49.1%。80例有细胞遗传学异常的病例中,t(8;21)异常58例,其中有2例为t(8;21)的复杂变异易位,1例为45,X,-X,t(2;8;21),另1例为46,XX,4q+,t(6;8;21),均死于白血病复发。统计发现,伴性染色体缺失的28例患儿均同时伴有t(8;21)平衡易位或变异易位。 2.2 各组患儿临床资料及近期疗效
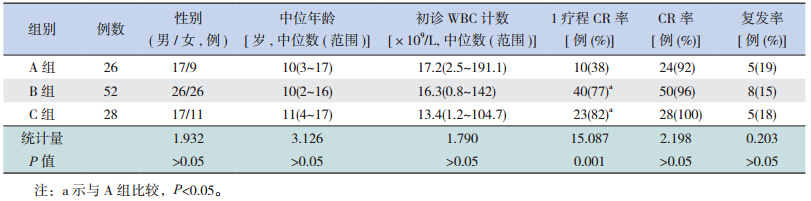
各组患儿在初诊WBC计数、完全缓解(CR)率和复发率上比较差异均无统计学意义,但是在1疗程CR率上,B、C组均明显高于A组(均P<0.05),见表 1。
| 表 1 各组患儿临床特征和近期疗效比较 |
采用Kaplan-Meier法进行生存分析表明,A、B、C组患儿中位EFS期分别为57.6、53.5和90个月;5年EFS率分别为38.9%±11.2%、59.3%±7.3%和66.5%±10.5%,其中C组5年EFS率明显高于A组(P=0.035),而其他组间两两比较差异均无统计学意义(P>0.05,图 1);A、B、C组患儿5年OS率分别为54.3%±13.5%、68.1%±7.7%和77.9%±9.8%,差异无统计学意义(P>0.05,图 2)。
 | 图 1 各组患儿无事件生存曲线 |
 | 图 各组患儿总生存曲线 |
根据性染色体缺失的不同将C组患儿分为X染色体缺失组(-X组,n=11)和Y染色体缺失组(-Y组,n=17),分别比较-X组和-Y组患儿与A组和B组在EFS率和OS率上的差异,结果显示,除-X组的EFS率明显高于A组外(P=0.033),其余各组间差异均无统计学意义(均P>0.05)。 2.5 伴t(8;21)易位的患儿性染色体缺失与预后的关系
伴t(8;21)易位的AMLM2型患儿共58例,其5年EFS率为63.3%±7.3%,明显高于正常核型患儿(38.9%±11.2%,P=0.015)。其中伴性染色体缺失的有28例(48%);不伴性染色体缺失的30例(52%),两组间EFS率比较差异无统计学意义(P>0.05)。根据细胞遗传学结果将58例患儿分为:单纯t(8;21)18例,占31%;t(8;21)伴性染色体缺失22例,占38%;t(8;21)伴其他附加染色体异常12例,占21%;t(8;21)伴性染色体缺失同时伴其他附加染色体异常6例,占10%,4组间EFS率比较差异无统计学意义(P>0.05)。 3 讨论
目前国际上根据预后等级将AML的细胞遗传学改变分为预后良好、预后中等和预后差三类染色体核型[5,6,7],其中没有提到性染色体缺失。本研究发现C组M2型患儿的1疗程CR率明显高于A组,5年EFS率也明显高于A组,提示性染色体缺失是AML预后好的染色体核型。
文献报道,伴t(8;21)异常的AML患儿性染色体缺失的频率远高于其他AML亚型,二者之间的关系至今仍不清楚[8]。本研究也发现性染色体缺失的病例均伴有t(8;21)的遗传学异常,由于文献[9]及本研究均证实伴有t(8;21)遗传学异常的AML患儿预后好于正常核型患儿,因此我们推测性染色体缺失的遗传学异常之所以预后好可能与他们同时伴有t(8;21)有关。
本研究中,t(8;21)异常的58例患儿伴性染色体缺失的有28例,发生率为48%;不伴性染色体缺失的30例,发生率为52%,两组间EFS率差异无统计学意义;将58例患儿根据细胞遗传学异常分为单纯t(8;21)、t(8;21)伴性染色体缺失、t(8;21)伴其他附加染色体异常和t(8;21)伴性染色体缺失同时伴其他附加染色体异常4组,4组间EFS率差异也无统计学意义;提示在伴有t(8;21)细胞遗传学异常的患者中,性染色体缺失并没有显示出更好的预后。这与大部分文献报道一致[10,11],但亦有少部分文献认为t(8;21)异常的AML性染色体缺失是预后好的标志[7,12,13]。Grimwade等[14]认为-X异常无预后价值,-Y异常提示预后较好,这与本研究相反,故不同染色体缺失与预后的关系需要大规模长期随访临床研究进一步确认。
本研究中有两例t(8;21)的复杂变异易位,1例为45,X,-X,t(2;8;21);另1例为46,XX,4q+,t(6;8;21),均死于白血病复发,提示t(8;21)的复杂变异易位不管是否具有性染色体丢失,可能预后不好。迄今为止文献报道的t(8;21)复杂变异易位共50余例,其中累及1号染色体的t(8;21)复杂变异易位最多见[15]。复杂变异易位与预后的关系不清楚,有文献报道该类患者对诱导化疗的反应不如典型t(8;21),预后较差,中大剂量Ara-C等强烈化疗能逆转这种不良预后[16]。
总之,本研究发现儿童AMLM2型的性染色体缺失是预后好的染色体核型,该类型几乎同时伴有t(8;21),在伴有t(8;21)细胞遗传学异常的患儿中,性染色体缺失并没有显示出更好的预后,推测性染色体缺失的遗传学异常之所以预后好可能与他们同时伴有t(8;21)有关。
| [1] | Rubnitz JE, Inaba H. Childhood acute myeloid leukaemia[J]. Br J Haematol, 2012, 159(3): 259-276. |
| [2] | Braoudaki M, Tzortzatou-Stathopoulou F. Clinical cytogenetics in pediatric acute leukemia: an update[J]. Clin Lymphoma Myeloma Leuk, 2012, 12(4): 230-237. |
| [3] | 竺晓凡. 如何提高儿童急性髓系白血病的疗效[J]. 中国当代儿科杂志, 2014, 16(2): 108-110. |
| [4] | 张之南, 沈悌. 血液病诊断及疗效标准[M]. 第2版. 北京: 科学出版社, 1999: 214-218. |
| [5] | Mrozek K, Marcucci G, Nicolet D, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia[J]. J Clin Oncol, 2012, 30(36): 4515-4523. |
| [6] | Kumari P, LinqappaKacitha B, Obula Reddy C, et al. A rare cytogenetic presentation of acute myeloid leukemia(AML-M2)[J]. Acta Med Iran, 2012, 50(12): 827-830. |
| [7] | 阮敏, 王雅琴, 张梦, 等. 儿童急性髓系白血病细胞遗传学改变与预后分析[J]. 中华血液学杂志, 2012, 33(9): 725-728. |
| [8] | 牧启田, 陈志妹, 楼基余, 等. 154例t(8;21)急性髓系白血病遗传学分析[J]. 浙江大学学报(医学版), 2010, 39(3): 236-240. |
| [9] | Estey EH. Acute myeloid leukemia: 2013 update on risk-stratification and management[J]. Am J Hematol, 2013, 88(4): 318-327. |
| [10] | Appelbaum FR, Kopecky KJ, Tallman MS, et al. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations[J]. Br J Haematol, 2006, 135(2): 165-173. |
| [11] | Lin P, Chen L, Luthra R, et al. Acute myeloid leukemia harboring t(8;21)(q22;q22): a heterogeneous disease with poor outcome in a subset of patients unrelated to secondary cytogenetic aberrations[J]. Mod Pathol, 2008, 21(8): 1029-1036. |
| [12] | Hann IM, Webb DK, Gibson BE, et al. MRC trials in childhood acute myeloid leukaemia[J]. Ann Hematol, 2004, 83(Suppl 1): S108-S112. |
| [13] | Rege K, Swansbury GJ, Atra AA, et al. Disease features in acute myeloid leukemia with t(8;21)(q22;q22). Influence of age, secondary karyotype abnormalities, CD19 status, and extramedullary leukemia on survival[J]. Leuk Lymphoma, 2000, 40(1-2): 67-77. |
| [14] | Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials[J]. Blood, 2010, 116(3): 354-365. |
| [15] | 何亚香, 薛永权, 王红英, 等. 伴有t(8;21)变异易位的急性髓系向血病患儿二例遗传学研究[J]. 白血病·淋巴瘤, 2012, 21(9): 517-519. |
| [16] | 戴海萍, 薛永权, 潘金兰, 等. 一例伴t(1;2l;8)(p36;q22;q22)复杂易位的急性髓系白血病的荧光原位杂交研究[J]. 中华血液学杂志, 2008, 29(8): 557-559. |