 2015, Vol. 17
2015, Vol. 17



50只SPF级健康BALB/c雌性小鼠,6~8周龄,体重20±2g,购于河南省实验动物中心。1,25-(OH)2D3和鸡卵清蛋白(ovalbumin,OVA)购于Sigma公司,免疫组化一抗购于北京博奥森生物技术有限公司,二抗及显色剂购于北京中杉金桥生物技术有限公司,Trizol、反转录试剂盒和PCR试剂购于Transgene公司。 1.2 动物分组和哮喘小鼠模型的制备
参照文献并加以改进制备动物模型[5]:适应性饲养1周后,按随机原则将50只BALB/c小鼠随机分为对照组、哮喘组、低剂量组、中剂量组和高剂量组,每组10只。在第1、8、15天哮喘组、低剂量组、中剂量组和高剂量组小鼠腹腔内注射抗原混合液0.2mL(含10%氢氧化铝0.15mL、OVA50μg和生理盐水0.05mL)致敏,对照组给予0.2mL生理盐水腹腔注射。第22天开始,将哮喘组、低剂量组、中剂量组和高剂量组小鼠置于自制的透明密闭容器内,由雾化器提供雾化动力,以1%OVA进行雾化吸入激发,每日1次,每次30min,持续至第35天。低剂量组、中剂量组、高剂量组在每次激发前30min分别按1、4、10μg/kg给予腹腔注射1,25-(OH)2D3混合液0.02mL[含1,25-(OH)2D30.02μg、无水乙醇0.5μL和生理盐水0.02mL]、0.08mL[含1,25-(OH)2D30.08μg、无水乙醇2.0μL和生理盐水0.08mL]、0.2mL[含1,25-(OH)2D30.2μg、无水乙醇5.0μL和生理盐水0.2mL];哮喘组每次雾化激发前30min给予腹腔注射生理盐水0.08mL;对照组雾化前30min腹腔注射和雾化激发均用生理盐水替代。 1.3 肺组织标本制备
各组小鼠于末次雾化激发结束后24h内以乙醚吸入麻醉,开胸,结扎左肺门,取出左肺,冻存于液氮中,用于RT-PCR检测;经右心室插管至肺动脉,用生理盐水快速冲洗至无血液流出,肺叶颜色呈白色后,换4%甲醛溶液冲洗,进行内固定,取出右肺,置于4%甲醛溶液中进行外固定72h,酒精梯度脱水,采用连续切片,3μm厚度切片,每隔3张切片选取1张,用于苏木精-伊红(HE)染色及免疫组织化学染色。 1.4 HE 染色及免疫组化染色观察
每个标本随机取3张切片,每张切片随机选取5个以上高倍镜视野;HE染色下,观察支气管壁的形态学改变,测定相同级别支气管横断面气道壁厚度;免疫组化染色下,应用实验室图像分析系统在高倍镜视野(10×40)下观察HMGB1及IL-17的表达。 1.5 RT-PCR 检测 HMGB1 及 IL-17 mRNA 表达
采用Trizol提取左肺组织总RNA,使用逆转录试剂盒合成cDNA,设计引物,扩增目的基因。HMGB1序列:上游5'-GAAGAGGAGGAAGAAGAGGA-3',下游5'-GCAAGGTTAGTGGCTATTGA-3',产物长度270bp;IL-17序列:上游5'-GAGAAGATGCTGGTGGGTGT-3',下游:5'-TTTCATTGTGGAGGGCAGAC-3',产物长度208bp;内参GAPDH序列:上游5'-GAGGCCGGTGCTGAGTATGT-3',下游5'-GGTGGAAGAGTGGGAGTTGCT-3',产物长度618bp。反应条件:94℃预变性2min;94℃变性30s,55℃退火30s,72℃延伸2min,共35个循环;72℃再延伸6min。扩增产物在2%琼脂糖凝胶上进行电泳,并采用凝胶电泳成像系统观察目的基因,目的基因mRNA的表达量以目的基因的DNA条带与内参基因的DNA条带灰度值的比值表示。 1.6 统计学分析
采用SPSS17.0统计软件对数据进行统计学分析,计量资料用均数±标准差(x±s)表示,多个样本均数间的比较采用单因素方差分析,组间数据的两两比较采用SNK-q检验;两个变量的相关分析采用Pearson相关分析。P<0.05为差异有统计学意义。2 结果 2.1 小鼠肺组织病理形态学观察
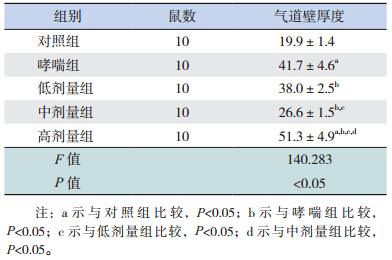
在普通光学高倍显微镜(10×40)下观察小鼠肺组织病理形态学变化。与对照组相比,哮喘组气道壁明显增厚(P<0.05),上皮细胞排列紊乱、脱落,气道周围有大量炎性细胞浸润;低剂量组和中剂量组的气道壁厚度明显低于哮喘组(P<0.05),且中剂量组气道壁厚度明显低于低剂量组(P<0.05);但高剂量组的气道壁厚度均明显高于其他4组(均P<0.05)。见图 1,表 1。
 | 图 1 各组小鼠肺组织病理形态学变化(苏木精 - 伊红染色,×400) 对照组小鼠支气管壁结构完整光滑,上皮细胞排列整齐,气道壁厚度适中,少量炎性细胞浸润;哮喘组小鼠支气管壁增厚受损、管腔狭窄,上皮细胞排列紊乱、脱落,支气管周围较多炎性细胞浸润;低剂量组小鼠支气管可见上皮细胞和平滑肌轻度增生,基底膜稍增厚,支气管周围可见少量炎性细胞浸润;中剂量组小鼠管壁增厚不明显,支气管周围可见较少的炎性细胞浸润;高剂量组可见支气管出现管壁明显增厚受埙,上皮细胞紊乱,周围大量炎性细胞浸润。 |
| 表 1 各组小鼠气道壁厚度 (x±s,μm) |
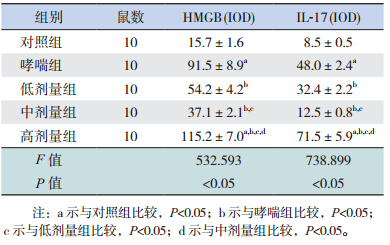
HMGB1和IL-17蛋白均主要表达于炎症细胞及上皮细胞的细胞核、胞浆。HMGB1、IL-17在哮喘组的表达明显高于对照组(P<0.05);HMGB1、IL-17在低剂量组、中剂量组的表达明显低于哮喘组(P<0.05),且中剂量组HMGB1、IL-17的表达明显低于低剂量组(P<0.05);但高剂量组中HMGB1、IL-17的表达均高于其他4组(均P<0.05)。见图 2,表 2。
 | 图 2 各组小鼠 HMGB1 和 IL-17 在肺组织内的表达(免疫组织化学染色,×400) 深棕黄色为 HMGB1 和IL-17 表达阳性细胞。对照组 HMGB1 和 IL-17 表达最弱;哮喘组 HMGB1 和 IL-17 呈强阳性表达;低剂量组 HMGB1 和 IL-17表达较哮喘组降低;中剂量组 HMGB1 和 IL-17 表达较低剂量组降低;高剂量组 HMGB1 和 IL-17 表达较哮喘组升高。 |
| 表 2 各组小鼠 HMGB1 及 IL-17 表达水平比较 (x±s) |
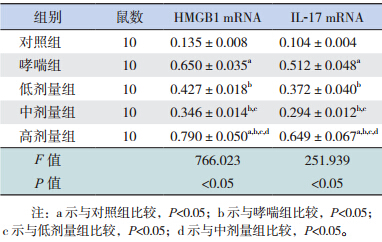
RT-PCR检测结果显示:HMGB1和IL-17mRNA在哮喘组的表达明显高于对照组(P<0.05);HMGB1和IL-17mRNA在低剂量组、中剂量组的表达明显低于哮喘组(P<0.05),且中剂量组HMGB1和IL-17mRNA的表达明显低于低剂量组(P<0.05);但在高剂量组中HMGB1和IL-17mRNA的表达均高于其他4组(均P<0.05)。见图 3、表 3。
 | 图 3 各组小鼠肺组织 HMGB1 及 IL-17 mRNA 的表达 注:M:Marker;1:对照组;2:低剂量组;3:中剂量组;4:哮喘组;5:高剂量组。 |
| 表 3 各组小鼠 HMGB1 及 IL-17 mRNA 表达 (x±s) |
肺组织内HMGB1蛋白的表达与气道壁厚度呈正相关(r=0.924,P<0.01),肺组织内IL-17蛋白的表达与气道壁厚度亦呈正相关(r=0.954,P<0.01);肺组织内HMGB1蛋白与IL-17蛋白的表达呈正相关(r=0.967,P<0.01),肺组织内HMGB1mRNA与IL-17mRNA的表达亦呈正相关(r=0.975,P<0.01)。见图 4~7。
 | 图 4 小鼠肺组织内 HMGB1 蛋白表达与气道壁厚度关系的散点图 |
 | 图 5 小鼠肺组织内 IL-17 蛋白表达与气道壁厚度关系的散点图 |
 | 图 6 小鼠肺组织内 HMGB1 蛋白与 IL-17 蛋白表达关系的散点图 |
 | 图 7 小鼠肺组织内 HMGB1 mRNA 与 IL-17 mRNA表达关系的散点图 |
哮喘是一种由免疫异常而导致反复发作的变态反应性疾病,已经证明Th17细胞及其细胞因子参与了气道炎症、气道重塑及气道高反应性,在哮喘中发挥着重要作用[6]。而IL-17作为一种强大的招募中性粒细胞因子,其在哮喘中的作用也相应逐渐受到重视。IL-17为Th17细胞的主要功能因子,由激活的T细胞产生,在中性粒细胞的募集、活化和迁移过程中发挥重要作用,诱导T细胞介导的慢性炎症反应。本研究发现哮喘组小鼠支气管及血管周围炎性细胞浸润显著增多,IL-17的表达明显高于对照组,提示IL-17可能参与了哮喘的发病过程。HMGB1作为一种晚期炎症因子,参与多种细胞因子、血管黏附分子、促炎细胞趋化等相互作用引起的炎症反应及信号转导过程[3]。有研究表明,哮喘患者痰中的HMGB1浓度明显升高[7],本研究也发现HMGB1在哮喘组中的表达显著升高。HMGB1能诱导树突细胞成熟并分泌多种促炎细胞因子[8],Anti-HMGB1中和抗体可通过抑制树突状细胞介导Th17极化改善中性气道炎症[9],提示HNGB1可作为炎症因子,直接参与哮喘的发病过程或作用于树突细胞,介导Th17细胞分化,间接参与哮喘的发病过程。He等[10]研究发现HMGB1通过对TLR2和IL-23表达的影响,参与Th17细胞分化。本研究发现,肺组织内HMGB1与IL-17表达呈正相关,提示HMGB1还可通过与Toll样受体(TLRs)等受体结合,介导IL-17的产生,参与哮喘气道炎症及气道重塑。1,25-(OH)2D3除发挥调节机体钙、磷代谢的生理功能外,还可通过与VDR结合,参与多种细胞的增殖和分化,参与免疫调节作用及多种肿瘤的发生过程[11]。VDR广泛分布于全身各处[12],几乎表达于所有免疫细胞,包括活化的CD4+、CD8+、T细胞、B细胞、单核细胞、中性粒细胞和抗原递呈细胞[13]。有研究证明HMGB1参与Th17细胞分化,介导IL-17的产生,1,25-(OH)2D3也可直接调节Th17细胞的分化,抑制IL-17等细胞因子表达[14,15],提示1,25-(OH)2D3可通过与VDR结合直接或间接参与对哮喘的调节作用[16]。本文研究发现,HMGB1、IL-17在哮喘组的表达明显高于对照组,而在低剂量组、高剂量组中的表达明显低于哮喘组。研究还发现肺组织内HMGB1与IL-17表达呈正相关,提示1,25-(OH)2D3可能通过降低哮喘小鼠肺内HMGB1的表达或阻断HMGB1-IL-17通路或直接下调Th17的数量和功能,减轻哮喘小鼠气道炎症。研究证明[11,17],1,25-(OH)2D3不仅能通过抑制HMGB1、TLR4及转化生长因子β1表达,还能通过直接抑制气道平滑肌细胞的增殖,减轻哮喘气道重塑。本研究发现,低剂量组、中剂量组的气道壁厚度明显低于哮喘组,说明1,25-(OH)2D3还可通过直接抑制气道平滑肌增殖,减轻气道重塑。Matheu等[18]研究发现,过量补充维生素D能促进IL-4、IL-13等细胞因子产生,加重气道重塑。本研究也发现高剂量组的气道壁厚度明显高于哮喘组,中剂量组气道壁厚度明显低于低剂量组,提示适量补充1,25-(OH)2D3可有效改善气道重塑,过量1,25-(OH)2D3可能加重气道重塑。本次试验研究表明,HMGB1及IL-17可能参与了哮喘气道重塑过程,不同剂量1,25-(OH)2D3能影响哮喘小鼠气道重塑,适量补充1,25-(OH)2D3不仅能通过降低哮喘小鼠肺内HMGB1的表达或阻断HMGB1-IL-17通路或直接下调Th17的数量和功能,还能直接抑制气道平滑肌细胞的增殖,减轻哮喘小鼠气道重塑,过量1,25-(OH)2D3能加重气道重塑,为进一步对哮喘发病机制的研究以及哮喘的治疗提供了新的认识。
| [1] | Braman S. The global burden of asthma[J]. Chest, 2006, 130(1 Suppl): 4S-12S. |
| [2] | Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex,heterogeneous disease[J]. Lancet, 2008, 372(9643): 1107-1119. |
| [3] | 张婷, 夏敏. 高迁移率族蛋白B1信号转导通路的研究进展[J]. 医学综述, 2011, 17(2): 195-198. |
| [4] | 王晨宇, 王星, 王琳, 等. Th17细胞分化调节机制及与自身免疫性疾病关系研究进展[J].细胞与分子免疫学杂志, 2014, 30(6): 660-662. |
| [5] | 王亚哲, 栾斌, 张艳丽, 等. 1,25-(OH)2D3对哮喘小鼠肺内TIM-4表达的影响[J]. 中国当代儿科学杂志, 2013, 15(1): 68-70. |
| [6] | Heelings PW, Kasran A, Liu Z, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma[J]. Am J Respir Cell Mol Biol, 2003, 28(1): 42-50. |
| [7] | Zhang F, Huang G, Hu B, et al. Anti-HMGB1 neutralizing antibody ameliorates neutrophilic airway inflammation by suppressing dendritic cell-mediated Th17 polarization[J]. Mediators Inflamm, 2014, 2014: 257930. |
| [8] | Wang YH, Liu YJ. The IL-17 cytokine family and their role in allergic inflammation[J]. Curr Opin Immunol, 2008, 20(6): 697-702. |
| [9] | Shim EJ, Chun E, Lee HS, et al. The role of high-mobility group box-1 (HMGB1) in the pathogenesis of asthma[J]. Clin Exp Allergy, 2012, 42(6): 958-965. |
| [10] | He Z, Shotorbani SS, Jiao Z, et al. HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis[J]. Scand J Immunol, 2012, 76(5): 483-490. |
| [11] | 谷惠如, 栾斌, 乔俊英, 等. 1,25-(OH)2D3对哮喘小鼠肺内高迁移率族蛋白B1及Toll样受体4表达的影响[J]. 中国当代儿科学杂志, 2014, 16(3): 301-305. |
| [12] | Hewison M. Vitamin D and immune function: an overview[J]. Proc Nutr Soc, 2012, 71(1): 50-61. |
| [13] | Holick MF, Chen TC. Vitamin D deficiency: a worldwide problem with health consequences[J]. Am J Clin Nutr, 2008, 87(4): 1080S-1086S. |
| [14] | Ryz NR, Patterson SJ, Zhang Y, et al. Active vitamin D (1,25-dihydroxyvitamin D3) increases host susceptibility to citrobacter rodentium by syppressing mucosal Th17 responses [J]. Am J Physiol, 2012, 303(12): 1299-1311. |
| [15] | Colin EM, Asmawidjaja PS, van Hamburg JP, et al. 1,25-dihydroxyvitamin D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis[J]. Arthritis Rheum, 2010, 62(1): 132-142. |
| [16] | Giarratana N, Penna G, Amuchastegui S, et al. A vitamin D analog down-regulates proinflammatory chemokine production by pancreatic islets inhibiting T cell recruitment and type 1 diabetes development[J]. J Immunol, 2004, 173(4): 2280-2287. |
| [17] | 宋颖芳, 赖国祥, 柳德灵, 等. 1,25-二羟基维生素D3抑制慢性哮喘模型小鼠肺组织α-平滑肌肌动蛋白的表达及气道重塑[J]. 实用医学杂志, 2012, 28(21): 3517-3520. |
| [18] | Matheu V, Back O, Mondoc E, et al. Dual effects of vitamin D-induced alteration of TH1/TH2 cytokine expression: Enhancing IgE production and decreasing airway eosinophilia in murine allergic airway disease[J]. J Allergy Clin Immunol, 2003, 112(3): 585-592. |