 2015, Vol. 17
2015, Vol. 17

2. 泸州医学院附属医院 医学实验研究中心, 四川 泸州 646000

 , LI Qing-Ping1, LEI Xiao-Ping1, KANG Lan1, GUO Lin1, ZHAI Xue-Song1, WANG Sheng-Hui1, CHEN Feng2
, LI Qing-Ping1, LEI Xiao-Ping1, KANG Lan1, GUO Lin1, ZHAI Xue-Song1, WANG Sheng-Hui1, CHEN Feng2
新生儿(特别是早产儿)因呼吸系统疾病常 需要长时间吸入高浓度和/ 或高压力氧气,幸存者 常常发生肺部氧化应激损伤,遗留明显的肺发育 障碍和肺功能衰竭,严重影响患儿生活质量[1]。 研究表明,高氧性肺损伤的机理与氧化应激、细 胞因子、一氧化氮、中性粒细胞的过度活化及肺 泡表面活性物质变化等多种因素密切相关[2, 3, 4]。活 性氧簇(reactive oxgen species,ROS)常通过还原 型烟酰胺腺嘌呤二核苷酸磷酸(triphosphopyridine nucleotide,NADPH)或线粒体途径产生[5]。关于 PKCβ/P66Shc 是否参与高氧环境下肺泡上皮细胞线 粒体内ROS 的产生,国内外尚无文献报道。本实 验从发生凋亡的氧化应激信号通路(线粒体前途 径)来阐明高氧性肺损伤的机制,从而为减轻高 氧肺损伤寻找新的治疗靶点。
研究显示,在高氧环境下线粒体发生氧化应 激反应,产生大量ROS[6, 7, 8],而ROS 是凋亡发生的 上游因子,可触发细胞的凋亡发生[8]。P66Shc 是 哺乳动物生命周期相关蛋白,主要功能是调节细 胞氧化应激反应和调控生命周期,是近年来研究 细胞氧化应激的关键蛋白[9]。主要定位于细胞浆, 正常情况下处于失活状态,不影响线粒体功能。 本课题组的前期研究结果表明,在高氧刺激下, 首先活化 PKCβ,使胞浆内的 P66Shc 发生磷酸化, 进而被脯氨酰异构酶(Pin 1)识别发生异构化(Pin 1 介导P66Shc 的线粒体转位),在蛋白磷酸酶2A (protein phos phatase 2A,PP2A)的作用下去磷酸 化进入线粒体,活化的P66Shc 转移到线粒体呼吸 链酶复合体Ⅲ氧化细胞色素C 产生ROS/H2O2 [10, 11]。 可见PKCβ/P66Shc 是调节细胞氧化应激反应重要 的信号通路。本课题组的前期研究已经建立了高 氧诱导人肺泡Ⅱ型细胞(A549)损伤模型[10],已 证实了mitoKATP 通道开放剂二氮嗪具有降低Omi/ HtrA2 表达和A549 细胞凋亡率,从而减轻高氧诱 导的肺损伤的作用[12]。并且已经证明高氧诱导肺 泡上皮细胞损伤的过程中,PKCβ/P66Shc 氧化应 激通路介导了细胞的凋亡的过程[10]。本研究将在 此基础上进一步探讨高氧诱导线粒体产生ROS 的 机制是否与PKCβ/P66Shc 途径有关,LY333531 是 PKCβ 选择性抑制剂,其是否对高氧诱导的肺泡上 皮细胞具有保护作用,是否能减轻高氧诱导的肺 泡上皮细胞的损伤,国内外文献未见报道。 1 材料与方法 1.1 A549 细胞传代培养与分组
A549 细胞由泸州医学院附属医院中心实验 室提供;A549 细胞复苏后,加入含10% 胎牛血 清(天津市灏洋生物制品科技有限责任公司,中 国) 的DMEM(H) 培养液(Hyclone 公司,美 国),置于37 ℃、5% CO2、饱和湿度的培养箱 中培养。待细胞生长达对数生长期时,在生物安 全柜FORMA1285( 美国Thermo) 中进行操作, 以0.25% 胰蛋白酶(碧云天公司)消化,传代接 种于50 mL 的细胞培养瓶中,随机分为对照组、 高氧组和LY333531 组。对照组仍置于37℃、5% CO2 培养箱中;高氧组换液后,以3 L/min 流速 通入950 mL/L 的O2 和50 mL/L 的CO2 高纯混合 气,10 min 后密闭培养;LY333531 组用终浓度为 10 μmol/L 的等量PKCβ 抑制剂LY333531 培养液 (美国Alexis 公司)预处理24 h 后,再用高氧诱 导A549 细胞,10 min 后密闭培养,24 h 后收获细胞。 其中高氧组在培养结束时用ML-IICB 数字智能测 氧仪(北京航天鹏诚仪器仪表有限公司)检测培 养瓶中氧浓度,低于90% 的标本弃去。 1.2 Western blot 检测细胞PKCβ、Pin1、 P66Shc 和P66Shc-Ser36 的表达
Western blot 采用聚丙烯酰胺凝胶电泳,被检 测物是蛋白质,“探针”是抗体(均购自ABcam 公司, 美国),“显色”用标记的二抗。经过SDS-PAGE分离的蛋白质样品,转移到固相载体(NC 膜)上, 以固相载体上的蛋白质或多肽作为抗原,与对应 的抗体起免疫反应,再与酶或同位素标记的第二 抗体起反应,经过底物显色或放射自显影以检测 蛋白水平的表达。每组重复6 次,用UVP 凝胶图 象处理系统Labworks4.6 软件分析目的条带灰度值。 1.3 激光共聚焦显微镜检测P66Shc 在线粒体内的转位
收集各组细胞,采用“MitoTracker”作为分子 探针标记线粒体(绿色荧光),特异性的抗-P66Shc 的荧光抗体标记P66Shc(红色荧光),用激光共 聚焦显微镜观察P66Shc 蛋白在线粒体内的表达情 况。发射波长为510 nm 及580 nm。每组重复8 次, 计算转位率。 1.4 激光共聚焦显微镜检测线粒体ROS 产生
MitoSox Red 是一种高选择性的检测线粒体内 ROS 的荧光染料,可自由透过活细胞膜进入细胞 内并选择性靶向线粒体,被线粒体内的超氧阴离 子ROS 氧化产生红色荧光。用激光共聚焦显微镜 检测红色荧光反映线粒体ROS 产生的情况。发射 波长为510 nm 及580 nm。使用Image-pro 6.0 图像 分析软件分析图像。每组重复8 次,确定平均光 密度(AOD),进行统计分析。 1.5 激光共聚焦显微镜检测线粒体膜电位的变化
线粒体膜电位(Ψm △)的测量采用JC-1 荧 光探针染细胞,按线粒体膜电位检测试剂盒(碧 云天公司,中国)说明操作。首先用JC-1 这种阳 离子染料对线粒体膜进行染色,再用激光共聚焦 显微镜检测。发射波长为530 nm 及590 nm。使用 Image-pro 6.0 图像分析软件分析图像。每组重复10 次,确定AOD,进行统计分析。 1.6 统计学分析
采用SPSS 18.0 统计软件对数据进行统计学分 析,计量资料用均数± 标准差(x±s)表示,多 个样本均数的比较采用单因素方差分析,组间两 两比较采用LSD-t 检验,P<0.05 为差异有统计学 意义。 2 结果 2.1 高氧对A549 细胞内PKCβ 表达及LY333531
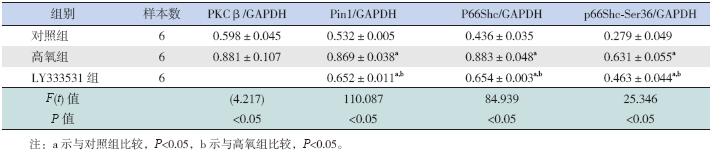
对高氧诱导A549 细胞内Pin1、P66Shc 和 P66Shc-Ser36 表达的影响 与对照组相比,高氧处理24 h 后A549 细胞 内PKCβ、Pin1、P66Shc 和P66Shc-Ser36 表达均 显著增加(均P<0.05);LY333531 处理组Pin1、 P66Shc和P66Shc-Ser36表达较高氧组均显著降低, 但仍高于对照组(均P<0.05)。见图 1,表 1。
 |
图 1 Western blot 条带图A: 高氧诱导下A549 细 胞内PKCβ 的表达;B:LY333531 对高氧诱导A549 细胞Pin1、 P66Shc 及P66Shc-Ser36 表达的影响。 |
| 表 1 各组细胞内PKCβ、Pin1、P66Shc 和P66Shc-Ser36 表达水平比较 (x±s) |
高氧组细胞P66Shc 线粒体转位率明显高 于对照组(P<0.05); 而LY333531 组较高氧组 P66Shc 线粒体转位率显著降低,但仍高于对照组 (P<0.05)。见图 2,表 2。
 |
图 2 LY333531 对高氧诱导A549 细胞P66Shc 在线粒体内转位的影响(激光共聚焦显微镜,×400)胞浆中线粒体绿色荧光与P66Shc 红色荧光重叠合成黄色荧光,代表P66Shc 已经发生转位;反之,胞浆中红色荧光与 绿色荧光未发生重叠而相互分离,代表P66Shc 未发生转位。对照组红绿荧光分离较多,代表P66Shc 在线粒体内的转 位率低,高氧组红绿荧光重叠,代表P66Shc 在线粒体内的转位率高,LY333531 组转位率较高氧组降低,但仍高于对 照组。 |
| 表 2 各组细胞P66Shc 线粒体转位率、ROS 产生和线粒体膜电位变化比较 (x±s) |
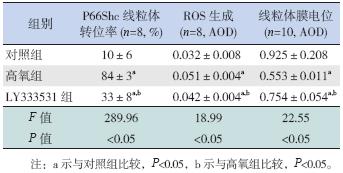
高氧组线粒体ROS 显著高于对照组(P<0.05), 并可见部分细胞核碎裂;而LY333531 组线粒体 ROS 较高氧组显著降低,但仍高于对照组(P<0.05)。 见图 3、表 2。
 |
图 3 LY333531 对高氧诱导A549 细胞线粒体ROS 产生的影响(激光共聚焦显微镜,×400)激光共聚 焦显微镜下线粒体ROS 呈红色荧光,细胞核呈蓝色荧光。高氧组红色荧光明显增多,代表线粒体ROS 显著高于对照组,而 LY333531 组线粒体ROS 较高氧组显著降低,但仍高于对照组。 |
高氧组细胞内的线粒体膜电位较对照组明显 降低(P<0.05);而LY333531 组线粒体膜电位较高氧组显著升高,但仍低于对照组(P<0.05)。见 图 4、表 2。
 |
图 4 LY333531 对高氧诱导A549 细胞线粒体膜电位的影响(激光共聚焦显微镜,×400)线粒体膜电位降 低时,在激光共聚焦显微镜下呈绿色荧光,线粒体膜电位增高时呈红色荧光,判断线粒体膜电位高低以红、绿荧光的比值来表示。 可见与对照组相比,高氧组线粒体膜电位降低,LY333531 组线粒体膜电位较高氧组有所升高,但仍低于对照组。 |
近年来,随着早产儿出生率的显著提高及呼 吸机的广泛使用,长期高浓度供氧气所引起的急、 慢性肺损伤是造成早产儿尤其是极低出生体重儿 死亡或致残的重要原因。研究表明,在高氧环境 下线粒体发生氧化应激反应,产生大量ROS 是其 发病的重要机制[7, 8, 13]。P66Shc 作为哺乳动物生命 周期相关蛋白,是近年来研究细胞氧化应激的关 键蛋白,其主要功能是调节细胞氧化应激反应和调控生命周期[9]。Pin1 是人类的肽基脯氨酰异构酶, 能特异地识别和结合蛋白质磷酸化的丝/ 苏- 脯氨 酸基序,催化磷酸化的丝/ 苏- 脯氨酸肽键发生顺 反异构,从而改变磷酸化蛋白质的功能[9, 14]。
本实验传代培养人肺泡上皮来源的A549 细胞 并建立高氧损伤细胞模型,主要探讨PKCβ/P66Shc 途径对线粒体的氧化损伤。结果显示,高氧处理 24 h 后,PKCβ、Pin1、P66Shc 和磷酸化P66Shc- Ser36 的蛋白表达量都明显高于对照组;且P66Shc 的转位率明显升高,ROS 产生增多,膜电位明显 下降,推测高氧诱导线粒体产生ROS 的机制与 PKCβ/P66Shc 途径有关。在高氧刺激下,首先活化 PKCβ,使胞浆内的 P66Shc 发生磷酸化,进而被 Pin1 识别发生异构化(Pin1 介导P66Shc 的线粒体 转位),在蛋白磷酸酶2A 的作用下去磷酸化进入 线粒体,活化的P66Shc 转移到线粒体呼吸链酶复 合体Ⅲ氧化细胞色素C 产生ROS/H2O2 [10, 11]。
氧化应激如何激活线粒体信号通路介导凋 亡发生?当ROS 超出其可承受的临界值后,便 诱导线粒体膜的渗透性改变和细胞色素C、Omi/ HtrA2 等的释放[6, 15]。凋亡因子被释放至细胞质, 它们或独立地破坏核内染色质,或激活凋亡蛋白 家族的主要成员- 胱冬肽酶,或作用于其他Ca2+ 依赖性蛋白,使整个细胞的结构破坏、功能紊 乱,最后细胞变成泡状凋亡小体而凋亡[16]。由此 可见PKCβ/P66Shc 氧化应激信号通路是作为线粒 体前途径介导了细胞的凋亡。同时,本实验采用 PKCβ 特异性抑制剂LY333531 阻断P66Shc 磷酸化 后,虽经过高氧干预,但A549 细胞中P66Shc 的 转位率(胞浆转移到线粒体)明显降低,ROS 产 生减少,膜电位明显有所上升,PKCβ 特异性抑 制剂LY333531 能明显抑制Pin1、P66Shc、磷酸 化P66Shc-Ser36 蛋白的表达。LY333531 其对氧化 应激状态下的A549 细胞具有的保护作用及其对 Pin1、P66Shc、P66Shc-Ser36 蛋白表达的影响更进 一步证明了PKCβ/P66Shc 氧化应激信号通路介导 高氧诱导肺泡上皮细胞内活性氧产生的作用。本 研究中LY333531 对高氧肺损伤的保护作用,为临 床治疗高氧肺损伤提供一定的理论依据。
| [1] | Gien J, Kinsella JP. Pathogenesis and treatment of bronchopulmonary dysplasia [J]. Curr Opin Pediatr, 2011, 23(3): 305-313. |
| [2] | Chetty A, Cao GJ, Manzo N, et al. The role of IL-6 and IL-11 in hyperoxic injury in developing lung [J]. Pediatr Pulmonol, 2008, 43(3): 297-304. |
| [3] | Ilizarov AM, Koo HC, Kazzaz JA, et al. Overexpression of manganese superoxide dismutase protects lung epithelial cells against oxidant injury [J]. Am J Respir Cell Mol Biol, 2001, 24(4): 436-441. |
| [4] | Gore A, Muralidhar M, Espey MG, et al. Hyperoxia sensing: from molecular mechanisms to significance in disease [J]. J Immunotoxicol, 2010, 7(4): 239-254. |
| [5] | Kardeh S, Ashkani-Esfahani S, Alizadeh AM. Paradoxical action of reactive oxygen species in creation and therapy of cancer [J]. Eur J Pharmacol, 2014, 735: 150-168. |
| [6] | Ratner V, Starkov A, Matsiukevich D, et al. Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice [J]. Am J Respir Cell Mol Biol, 2009, 40(5): 511-518. |
| [7] | Pagano A, Donati Y, Metrailler I, et al. Mitochondrial cytochrome c release is a key event in hyperoxia-induced lung injury: protection by cyclosporin A [J]. Am J Physiol Lung Cell Mol Physiol, 2004, 286(2): L275-283. |
| [8] | Waxman AB, Kolliputi N. IL-6 protects against hyperoxiainduced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusins [J]. Am J Respir Cell Mol Biol, 2009, 41(4): 385-396. |
| [9] | Lee TH, Pastorino L, Lu KP. Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease [J]. Expert Rev Mol Med, 2011, 13: e21. |
| [10] | 车忠丽, 董文斌, 李清平, et al. PKCβ/P66Shc 氧化应激通路 介导高氧诱导人肺泡上皮细胞凋亡的作用 [J]. 临床儿科杂 志, 2013, 31(11): 1066-1069. |
| [11] | Pinton P, Rimessi A, Marchi S, et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant P66Shc [J]. Science, 2007, 315(5812): 659-663. |
| [12] | 邹新艳, 董文斌, 邹丹, et al. mitoKATP 通道开放剂对高氧 诱导人A549 细胞凋亡的保护作用 [J]. 中国当代儿科杂志, 2011, 13(6): 514-517. |
| [13] | Ricci C, Pastukh V, Leonard J, et al. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis [J]. Am J Physiol Cell Physiol, 2008, 294(2): C413- 422. |
| [14] | Hanes SD. The Ess1 prolyl isomerase: traffic cop of the RNA polymerase II transcription cycle [J]. Biochim Biophys Acta, 2014, 1839(4): 316-333. |
| [15] | Delavallee L, Cabon L, Galan-Malo P, et al. AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics [J]. IUBMB Life, 2011, 63(4): 221-232. |
| [16] | Giorgio M, Migliaccio E, Orsini F, et al. Electron transfer between cytochrome c and P66Shc generates reactive oxygen species that trigger mitochondrial apoptosis [J]. Cell, 2005, 122(2): 221-233. |