 2015, Vol. 17
2015, Vol. 17



Fanconi-Bickel综合征(FBS, OMIM 227810)是一种罕见的糖代谢异常的常染色体隐性遗传病,主要表现为喂养困难、运动及体格发育落后、肝脏增大、多饮多尿、膝内翻或膝外翻、低血糖、高脂血症、高乳酸血症、低磷血症、肝功能异常、尿糖和尿蛋白阳性、高尿钙、高尿磷、代谢性酸中毒、骨质疏松等[1]。1997年Santer等[2]发现FBS致病基因为SLC2A2(GLUT2),该基因编码的葡萄糖转运载体GLUT2蛋白在肝脏、胰腺β细胞、肠细胞及肾小管细胞中表达[3]。SLC2A2位于染色体3q26.1-26.3,有11个外显子,长度30 kb[4]。据人类基因突变数据库(HGMD,http://www.hgmd.org)报道,到2014年4月FBS共有突变68种。
本研究通过对3例疑似FBS患儿及其家人进行SLC2A2基因分析,以明确致病突变,并总结其临床特征,提高对疾病表型的认识。
1 资料与方法 1.1 研究对象研究对象为2006~2012年于我院就诊的3例疑似FBS患儿(称为患儿A、B、C),均为女童,就诊年龄分别为30、16、21个月,随诊时间2~7年。其中患儿A曾被本课题组报道[5]。
临床拟诊标准:运动及体格发育落后、肝脏增大、伴或不伴佝偻病表现。实验室检查有空腹低血糖、高脂血症、高乳酸血症、低磷血症、尿糖和尿蛋白阳性、高尿钙、高尿磷和代谢性酸中毒等。
1.2 SLC2A2基因PCR扩增取患儿静脉抗凝外周血2 mL,应用北京博迈德科技发展公司柱型外周血基因组DNA提取试剂盒提取基因组DNA;应用Primer3在线设计引物(http://bioinfo.ut.ee/primer3-0.4.0),由英潍捷基(上海)贸易有限公司合成(引物序列见表 1),对SLC2A2全部11个外显子及内含子连接处进行PCR扩增。
| 表 1 SLC2A2 各外显子片段引物序列 |

PCR体系:博迈德® 2×PCR mix 12 μL,dd H2O 11 μL,DNA 1 μL,10 μmol/L引物1 μL。PCR 条件:95℃ 5 min;每个循环94℃ 30 s,58℃ 30 s,72℃ 45 s,共35个循环;72℃延伸10 min,4℃维持。PCR产物用2%西班牙琼脂糖(Biowest Agarose)凝胶电泳分离,切胶纯化,测序。检测到可疑突变位点后,对能提供标本的患儿父母进行相应位点检测。
1.3 基因变异致病性验证将检测到的变异通过HGMD及NCBI进行检索,对于未报道突变反向验证,并检测同意提供标本的患儿父母相应位点;未报道的错义突变应用SIFT(http://sift.jcvi.org)在线推测其致病性。
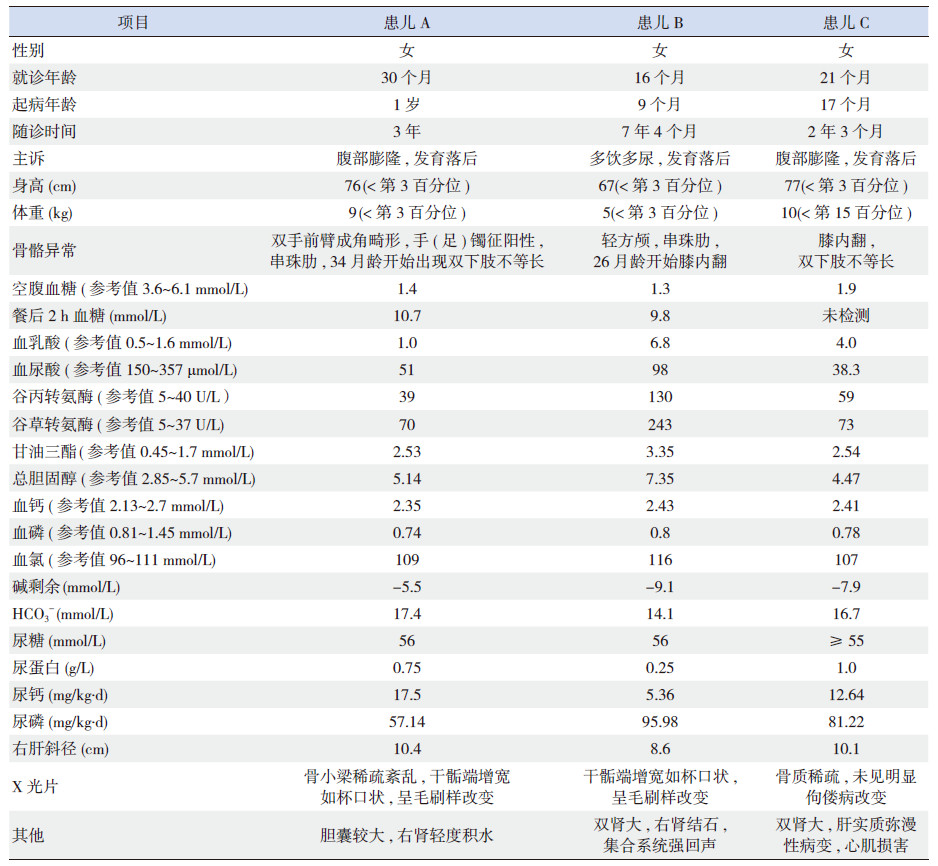
2 结果 2.1 临床表现3例患儿中2例主诉为“腹部膨隆,发育落后”,另1例为“多饮多尿,发育落后”。患儿均存在体格发育迟缓(身高均位于第3百分位以下,2例体重低于第3百分位)。运动发育落后主要表现为独坐、爬和行走时间明显推迟。3例均有多饮多尿症状,体查均可见腹部膨隆、肝大、佝偻病体征以及不同程度的骨骼畸形。实验室检查可见空腹低血糖、餐后高血糖、高乳酸、高血脂、转氨酶增高、低血磷,血气分析示代谢性酸中毒、HCO3-降低;蛋白尿、大量糖尿、24 h尿钙和尿磷增高。腹部B超均示肝大,可有肾大、胆囊大、肾积水等。双膝关节、双腕关节或骨盆正侧位X线片示程度不等的佝偻病改变或骨质疏松。1例患儿血氯增高;1例出现了心肌损害(心电图示PR间期延长,T波各导联切迹;24 h动态心电图示窦性心律不齐,Ⅰ°房室阻滞,QT间期延长,ST段抬高,有时可见Ⅰ、Ⅱ、Ⅲ导联T波切迹)。所有患儿智力正常,血钙和尿酸无明显异常,脾脏不大。3例患儿的临床资料见表 2。
| 表 2 3 例患儿初诊临床资料 |
3例患儿分别发现了SLC2A2基因纯合或复合杂合突变,确诊为FBS。患儿A发现纯合剪切突变:IVS8+5G>C(c.1068+5 G>C),其母亲检测到杂合突变:IVS8+5G>C(c.1068+5 G>C),无父亲标本;患儿B发现纯合无义突变:c.1194T>A(p.Tyr398X),抱养儿,无亲生父母标本;患儿C 发现2个杂合突变:错义突变c.380C>A(p.Ala127Asp)及重复突变c.970dupT(p.324TyrfsX392),其父亲发现杂合突变c.380C>A(p.Ala127Asp),母亲未发现突变(图 1)。
 |
图 1 FBS 家庭基因突变测序图谱 3 例患儿均检测到纯合突变或复合杂合突变,其中患儿A 父亲及患儿B 父母 未收集到标本;患儿C 的突变c.970dupT(p.324TyrfsX392)并非来源于父母,为新生突变。箭头所指为突变位点。 |
反向测序结果显示这些变异真实存在,经HGMD检索及文献检索,除本课题组前期报道的剪切突变c.1068+5 G>C外,其余3种突变均未见报道。其中仅有c.380C>A(p.Ala127Asp)为错义突变,应用SIFT推测,SIFT值为0,结果显示其为有害性新错义突变。另c.1194T>A(p.Tyr398X)为无义突变,c.970dupT(p.324TyrfsX392)为移码突变,致病性明确。
3 讨论FBS于1949年最早由Fanconi 及 Bickel报道[6],患儿3~10个月时即可有发热、呕吐、生长发育延迟等,之后会出现空腹低血糖、矮小、腹膨隆、肝大、满月脸以及肩膀和腹部脂肪沉积这些类似于糖原累积症(GSD)的症状[1]。另外FBS具有典型的近端肾小管肾病表现,出现肾小管酸中毒、大量尿糖,其肾脏会随身高增长而增大。患儿常可见牙齿畸形、易位及牙釉质缺损等。低磷性佝偻病及骨质疏松最常见,有的甚至会导致骨折[1]。有的患儿未发现肾小管功能缺陷及身材矮小[1],肝肿大也不是FBS确诊的必要条件[7]。FBS患儿的预后都较好,部分患儿成年后病情稳定。2011年von Schnakenburg 等[8]报道了1例成功孕育的FBS患者。
本文所报道的3例患儿临床症状与国外报道基本符合。患儿身高体重多低于第3百分位,均有腹膨隆,肝大但未见脾大,骨骼畸形以四肢畸形为主。实验室检查空腹血糖可低至1.5 mmol/L左右,多有血乳酸升高,谷草转氨酶升高比谷丙转氨酶明显,血脂以甘油三酯升高为主,低血磷,血气分析示代谢性酸中毒;但血尿酸无增高,血钙正常,血氯升高不明显。所有患儿尿钙、尿磷、尿蛋白均阳性。影像学检查均可见肝大,1例有肝实质弥漫性病变,但均未见脾大,肾脏均有异常,可表现为肾大、积水及肾结石。1例胆囊大。所有患儿X片均提示为佝偻病或骨质疏松。除经典症状外,患儿C还出现了心肌损害。
可见FBS的临床表现多与GSD(尤其是Ia型)以及以近端肾小管酸中毒为主要表现的Fanconi综合征(FS)相重叠,需要与二者进行鉴别。GSD Ia 与FBS所不同的表现主要有:GSD Ia常有血小板功能障碍、高尿酸血症,而FBS通常没有。并且GSD Ia肾脏受累为远端肾小管功能失调,所以没有FBS的高尿糖、低磷性佝偻病等近端肾小管功能缺陷的表现。FS主要由于近端肾小管重吸收障碍而引起葡萄糖尿、蛋白尿、尿磷、尿钾、碳酸氢盐尿及氨基酸尿等。患者常伴有乏力、骨痛及尿崩导致的血容量不足[9]。FS通常不伴有低血糖及相关表现。FBS最初的症状具有隐匿性,不易被察觉,本研究中的3例患儿均在1岁左右起病,往往出现比较明显的症状(如腹部膨隆)后才就医,而此时患儿各系统已经明显受累。监测尿糖、血糖可能有助于该病的早期发现。
SLC2A2编码的GLUT2是低亲和力的单糖转运体,介导D-葡萄糖和少量D-半乳糖、D-甘露糖和D-果糖的转运 [10]。Glut2缺陷导致患者对葡萄糖及半乳糖等不耐受。因其无法有效转运,空腹时糖原降解产生的葡萄糖无法转运至胞外,而造成胞内葡萄糖浓度升高,肝糖原累积,血糖降低;餐后血糖不能有效转运至肝脏胞内形成糖原,表现为高血糖。糖原累积进而出现上述类似于GSD的症状。肾小管细胞GLUT2缺陷使对葡萄糖的重吸收降低,导致了大量的尿糖[1]。肾小管酸中毒,高尿糖,钙磷代谢异常导致的高尿钙、尿磷,进而导致骨质疏松、佝偻病等。Santer等[7]认为人群中该基因的突变率很低,在他们所发现的33种突变中没有突变热点。Su等[11]于2011年报道了2例中国FBS患者,分别检测到复合杂合突变c.682C>T(p.Arg228X)和 c.1185G>A(p.Trp395X),以及c.196G>T(p.Glu66X)和c.1117delA(p.Met373X),均为无义突变。
本研究共检测到4种突变:剪接位点突变IVS8+5G>C(c.1068+5 G>C),无义突变 c.1194T>A(p.Tyr398X),错义突变c.380C>A(p.Ala127Asp)及重复突变c.970dupT(p.324TyrfsX392)。除本课题组曾报道的 IVS8+5G>C(c.1068+5 G>C)外,其余3种均为新突变。结合Su等[11]所报道的2例中国病例,国人SLC2A2突变中无义突变最为常见,这与国外报道类似。本研究的3名患儿的临床表现没有明显的差异,未发现基因型与表型之间明确的相关性。患儿C为杂合复合突变,其中c.380C>A(p.Ala127Asp)来源于父亲,而c.970dupT(p.324TyrfsX392)并未在其父母亲发现,提示c.970dupT(p.324TyrfsX392)并非经父母遗传,而是患儿C自身发生了基因突变而致病,为新生突变。该突变位点为polyT区域,有可能在DNA复制过程中出现了识别错误导致多复制了一个T。迄今国内外未见该疾病有新生突变的报道,本研究报道可能为首例。无义突变c.1194T>A(p.Tyr398X)及移码突变c.970dupT(p.324TyrfsX392)均导致终止密码子提前出现,造成提前终止蛋白翻译产生截短蛋白或引起蛋白降解,是明确的致病突变。由于SLC2A2基因的mRNA在外周血中的表达量过低,本研究未完成对剪接突变c.1068+5 G>C所导致的RNA具体剪接结果的研究。在之后的研究中,如能获患者同意,我们会对mRNA表达含量高的组织或细胞进行进一步研究。
综上所述,本研究对3例FBS患儿进行了SLC2A2基因分析而确诊,并对患者的临床表现进行了概括总结。本研究发现了4种基因突变,除本课题组曾报道的1种剪切突变外,余3种为新突变,未发现基因型与表型之间明确的相关性。患儿C 发现了一个新生突变c.970dupT(p.324TyrfsX392),可能为国内外首例新生突变报道。
| [1] | Santer R, Schneppenheim R, Suter D, et al. Fanconi-Bickel syndrome—the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature[J]. Eur J Pediatr, 1998, 157(10): 783-797. |
| [2] | Santer R, Schneppenheim R, Dombrowski A, et al. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome[J]. Nat Genet, 1997, 17(3): 324-326. |
| [3] | Mueckler M, Kruse M, Strube M, et al. A mutation in the Glut2 glucose transporter gene of a diabetic patient abolishes transport activity[J]. J Biol Chem, 1994, 269(27): 17765-17767. |
| [4] | Takeda J, Kayano T, Fukomoto H, et al. Organization of the human GLUT2 (pancreatic beta-cell and hepatocyte) glucose transporter gene[J]. Diabetes, 1993, 42(5): 773-777. |
| [5] | 张乐嘉, 邱正庆, 丁国芳, 等. Fanconi-Bickel 综合征1 例并文献复习[J]. 实用儿科临床杂志, 2011, 26(24): 1882-1884. |
| [6] | Fanconi G, Bickel H. Die chronische Aminoacidurie (Aminosaeurediabetes oder nephrotisch-glukosurischer Zwergwuchs) bei der Glykogenose und der Cystinkrankheit[J]. Helv Paediat Acta, 1949, 4(5): 359-396. |
| [7] | Santer R, Groth S, Kinner M, et al. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi-Bickel syndrome[J]. Hum Genet, 2002, 110(1): 21-29. |
| [8] | von Schnakenburg C, Santer R. Fanconi-Bickel syndrome and fertility[J]. Am J Med Genet A, 2011, 155A(10): 2607. |
| [9] | Murphy N, Elramah M, Vats H, et al. A case report of deferasirox-induced kidney injury and Fanconi syndrome[J]. WMJ, 2013, 112(4): 177-180. |
| [10] | Leturque A, Brot-Laroche E, Le Gall M. GLUT2 mutations, translocation, and receptor function in diet sugar managing[J]. Am J Physiol Endocrinol Metab, 2009, 296(5): E985-E992. |
| [11] | Su Z, Du ML, Chen HS, et al. Two cases of Fanconi-Bickel syndrome: first report from China with novel mutations of SLC2A2 gene[J]. J Pediatr Endocrinol Metab, 2011, 24(9-10): 749-753. |