引用本文

周渊峰, 乔中伟, 王艺.4岁男孩进行性运动和智力障碍1年余[J]. 中国当代儿科杂志, 2015, 17(6): 577-579.

ZHOU Yuan-Feng, QIAO Zhong-Wei, WANG Yi . Motor and intellectual disorders for more than one year in a 4-year-old boy[J]. CJCP, 2015, 17(6): 577-579.
复旦大学附属儿科医院神经内科, 上海 201102
[收稿日期]2014-12-22;[接受日期]2015-03-11
作者简介:周渊峰, 男, 博士, 副教授。
Motor and intellectual disorders for more than one year in a 4-year-old boy
Department of Neurology, Children's Hospital, Fudan University, Shanghai 201102, China
病例介绍
患儿男,4 岁,因进行性运动、智力障碍 1 年
余就诊。该患儿系第 4 胎第 1 产,产钳助产出生,
出生体重 3 800 g,无窒息抢救史,15 月龄前发育
不详。15 月龄时可独立行走,2 岁 10 个月起出现
走路不稳,平时走路易摔倒、跌倒,并出现触电
样动作,2~3 次 / 周,可自己站起,语言发育明显
落后,只会无意识叫“爸妈”及理解简单对话。
当时来到我院神经科门诊第 1 次就诊。父母体健。
体格检查:头围 48 cm(<1 SD),四肢肌力、肌
张力正常,走路姿势未见明显异常,发育筛查测
试(DST) 检 查 发 育 商(DQ)<50; 智 商(MI)
<46,诊断为“发育迟缓”,在我院给予康复训练
等治疗,但患儿运动障碍无明显缓解并呈进行性
加重、下肢更加明显,同时出现智力和认知能力
的倒退。3 岁 3 个月发热时抽搐一次,表现为全面
强直 - 阵挛发作,在我院行睡眠脑电图检查未见
明显异常放电,未给予抗癫癎药物治疗。4 岁时因
为丧失行走能力来我院神经科门诊再次就诊。体
格检查:头围 49 cm(<1 SD ),反应迟钝,张口,
流涎,肝脾肋下未及,四肢肌力Ⅳ级,肌张力增高、
双侧腱反射亢进,巴氏征(+),皮肤未见牛奶咖
啡斑和色素脱失斑。血液生化和血气分析、血尿
串联质谱、染色体等检查均为阴性,睡眠脑电图
提示大量广泛性高波幅 1~2 Hzδ 波持续性发放,
后头部夹杂低 - 中波幅棘波和尖波(图 1)。骨骼
肌和皮肤活检中骨骼肌病理像经酸性磷酸酶染色显示肌纤维胞浆内大量深红色点状沉积物,提示
酸性磷酸酶活性显著增高(图 2)。
皮肤电镜图显示真皮组织内大量类圆形或短
杆状脂褐素沉积物,呈簇状聚集(图 3)。常规头
颅 MRI、磁共振波谱分析(MRS)和磁共振血管
成像检查提示全脑萎缩样改变,小脑更明显;双
侧丘脑神经元损伤可能,双侧基底节区可见少许
乳酸峰;颅内血管未见明显异常结构性改变(图 4)。
病例讨论
周主治医师(神经科):4 岁学龄前期儿童,
临床主要表现为:慢性进行性运动和智力倒退;
发病前全面发育落后;神经系统体查发现头围偏
小、病理征阳性和四肢轻度瘫痪;有癫癎发作和
脑电图异常放电;头颅影像学提示全脑皮质萎缩,
小脑最明显。首先从定位诊断上,结合该患儿病
史、体格和辅助检查的特点,应考虑大脑皮层和
骨骼肌同时受累;其次在定性诊断上,遗传代谢
性疾病或神经变性病应首先考虑,从遗传代谢性
疾病或神经变性病的诊断思路来说,首先应详细
询问病史并仔细进行神经系统体查,其次可考虑
常规辅助检查,如血尿常规、肝肾功能、电解质、
心肌酶谱、脑电图和头颅影像学等检查,再者应进一步常规代谢筛查,如血糖、血氨、血乳酸等
检查,最后在常规辅助和代谢检查的基础上,可
进一步考虑特殊检查,如血尿串联质谱、酶活性
测定、组织活检、基因分析等。因此,该患儿在
常规和一些特殊检查不能得出正确诊断的基础上,
结合该患儿定位诊断在大脑皮层和骨骼肌,应首
先考虑骨骼肌和皮肤活检。
乔教授(放射科):该患儿头颅影像学检查
显示全脑弥漫性灰白质萎缩样改变,小脑更明显,
临床应考虑线粒体脑病、神经元蜡样质脂褐质沉
积症(neuronal ceroid lipofuscinoses,NCLs)以及代
谢性脑病等疾病。
王教授(神经科):根据患儿临床表现、辅
助检查以及肌肉和皮肤病理的结果,NCLs 诊断明
确。NCLs 是一群具有遗传异质性的神经元退行性疾病,发病率约为 1 : 12 500~1 : 100 000,多为常染
色体隐性遗传,多数在儿童期发病。其临床主要
表现为发育迟缓、进行性智力障碍和运动障碍(痴
呆、不自主运动、共济失调和痉挛)、视力丧失
和癫癎。根据发病年龄和临床特征,NCLs 可分为
先天型(生后 ~1 个月发病)、婴儿型(6 个月 ~2
岁发病)、晚发婴儿型(2~4 岁发病)、青少年型(4~8
岁发病)和成人型(15 岁后发病),临床分型有
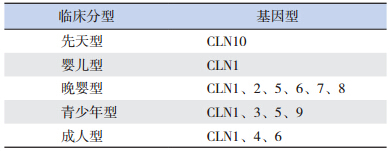
助于诊断及判断预后。依据致病基因定位,可分
为 10 种亚型,即 CLN1~10 型。基因型和临床分型
之间一般无直接相关性,如 CLN1 型基因突变可以
导致 4 种临床表型(表 1)[1]。NCLs 不同的基因型
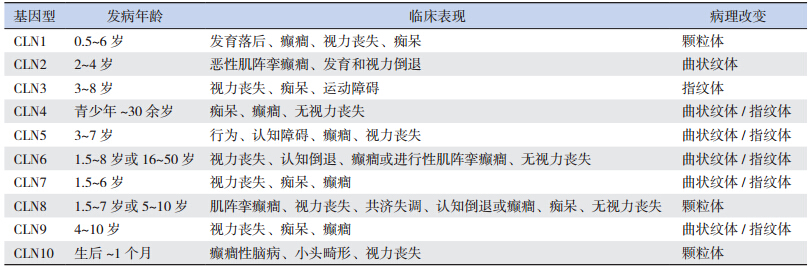
具有不同的临床表型(表 2)[2]。结合该患儿的发
病年龄,应考虑为晚发婴儿型。在辅助检查方面,
头颅影像学检查主要提示脑萎缩样改变,眼底检
查可有视神经萎缩,视觉诱发电位潜伏期延长、
波形缺如以及巨大视觉或体感诱发电位,脑电图
一般无特征性改变 [3],最终确诊主要依赖于电镜检
查、酶学分析和分子遗传学检查。采用电镜对全血、皮肤、结缔组织或其他组织标本进行脂褐素沉积
物的病理学检查是 NCLs 诊断和鉴别诊断的标准方
法,其病理改变主要有颗粒体沉积、曲状纹体沉
积和指纹体沉积,不同的基因型有不同的病理改
变(表 2)[2, 4]。此外,在临床上还可以对 CLN1 和
CLN2 的蛋白产物棕榈酰蛋白硫脂酶 1(PPT1)和
三肽基酶肽 1(TPP1)进行酶活性分析。分子遗传
学检查主要用于 NCLs 诊断、遗传咨询和产前诊断,
目前除 CLN9 型基因和染色体定位尚未明确外,其
他基因型基因和染色体定位已基本确定,可以利
用突变分析和二代测序技术进行检测。
表 1(Table 1)
 | 表 1 NCLs 临床分型和基因型之间的关系 |
表 2(Table 2)
| | 表 2 NCLs 不同基因型的临床表现和病理改变 |
 2015, Vol. 17
2015, Vol. 17