 2015, Vol. 17
2015, Vol. 17



先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH) 是 儿 科 较 常 见 的 常 染 色 体 隐 性遗传病,由肾上腺皮质激素合成途径中所需酶 的先天性缺陷所致。国外的数据表明,新生儿期 CAH 的 发 病 率 约 为 1/16 000~1/20 000[1]。常见的 CAH 分别由 21- 羟化酶、11B- 羟化酶、3β- 类固醇 脱氢酶、17- 羟化酶等缺陷引起。临床上常有性发 育异常和水、电解质紊乱(多为高血钾、低血钠)、 高血压等表现。合并有水、电解质紊乱的失盐型 患儿通常在婴幼儿期发病,为新生儿及儿科临床 上的急症之一,病情进展快、且最易误诊;而单 纯的性发育异常者因典型临床症状的缺失往往就 诊较迟。为加深儿科同行对本病的认识,现将本 院 2010~2014 年收治的 52 例 21- 羟化酶缺乏 CAH 患儿临床资料进行回顾性总结、现报告如下。 1 资料与方法 1.1 研究对象
选取 2010 年 9 月至 2014 年 9 月于本院新生 儿科及内分泌科住院治疗的 52 例 21- 羟化酶缺乏 CAH 患儿为研究对象。收集所有患儿的临床资料, 包括:性别、发病年龄、出生体重、家族史、临 床症状、体征、实验室检查结果、合并症、治疗 措施、随访情况等。根据病情轻重、是否伴随低 钠高钾酸中毒等失盐型表现,可分为单纯男性化 型(n=15)、失盐型(n=28)和非典型型(n=9) 3 组。 1.2 21- 羟化酶缺乏 CAH 诊断标准
诊断标准参考文献 [2]。
(1)临床表现:单纯男性化型:女性男性化 或男性假性性早熟,肤色深;失盐型:除以上表 现外尚有呕吐、腹泻、脱水、酸中毒及低钠、低氯、 高钾血症等表现;非典型型:无明显相关临床症状、 体征,实验室检测指标示皮质各激素水平异常。 (2)实验室检查:女性患儿染色体均为 46,XX, 血 17-α 羟孕酮明显升高,血皮质醇浓度正常或偏 低,睾酮升高,血管紧张素和醛固酮不同程度增高, 醛固酮在单纯男性化者可正常或偏低(激素检查 时间均在激素治疗前,促肾上腺皮质激素及皮质 醇均未检测节律)。血电解质示低血钠、高血钾、 酸中毒。 1.3 方法
52 例患儿进行了详细的体格检查,并检测了 血 17-α 羟孕酮、睾酮、雌二醇、促肾上腺皮质激 素(adrenal corticotropic hormone,ACTH)、血钾、 血钠水平及肾上腺影像学等;部分患儿对血气、 骨龄、染色体核型、突变基因等进行检测分析。 对于临床及实验室检测不典型的患儿,加做 ACTH 激发试验。治疗后定期随访,随访时间从 1 个月 至 4 年不等。大部分患儿确诊后均口服氢化可的 松(皮质功能危象者先静脉应用氢化可的松, 8~10 mg/kg,每日 2~3 次,待危象解除后改口服), 失盐型患儿加服食盐 1.5~3.0 g/d,严重失盐型患儿 加用 9-α 氟氢可的松 0.05~0.15 mg/d。 1.4 统计学分析
采用 SPSS 13.0 统计软件对数据进行分析,计 数资料采用百分率(%)表示;服从正态分布计量 资料采用均数 ± 标准差(x±s)表示,两组间比 较采用t检验;非正态分布计量资料采用中位数(四 分位间距)[P50(P25,P75)] 表示,两组间比较采 用 Mann-Whitney U 检验。P<0.05 为差异有统计学 意义。 2 结果 2.1 不同类型患儿一般资料
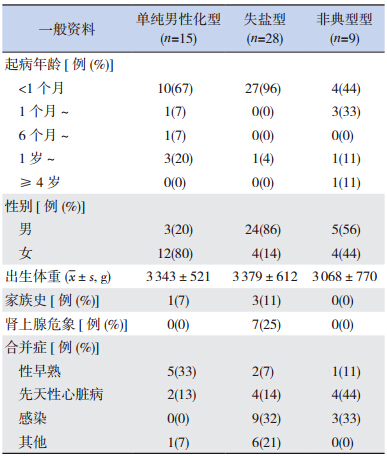
52 例 CAH 患 儿 中,男 32 例,女 20 例; 起 病年龄 1 d 至 13 岁 11 个月,其中 <1 个月 41 例、 1 个月 ~ 4 例,6 个月 ~ 1 例,1 岁 ~ 5 例,≥ 4 岁 1 例;出生体重 2 450~5 000 g;4 例有相关家族史, 其中 3 例兄弟患病、1 例姐弟患病。具体情况见表 1。
| 表 1不同类型患儿一般资料 |
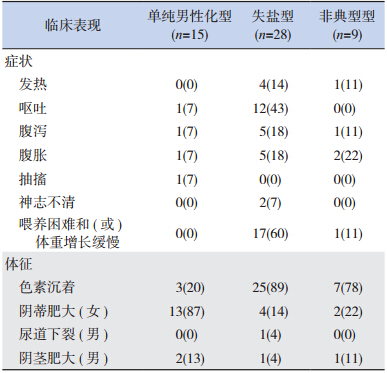
15 例单纯男性化型患儿中,有 1 例因呕吐、 腹泻、腹胀入院,1 例因抽搐入院;13 例表现为 阴蒂肥大,3 例有外阴色素沉着,2 例有阴茎肥大。 28 例失盐型患儿中,12 例因呕吐入院,5 例表现 出腹胀、腹泻,4 例有发热,2 例入院时存在神志 不清,17 例存在喂养困难和(或)体重增长缓慢; 25 例表现为色素沉着,4 例有阴蒂肥大,1 例有尿 道下裂,1 例有阴茎肥大。9 例非典型型患儿中, 2 例表现为腹胀,1 例表现为发热、腹泻,1 例表 现为体重增长缓慢;7 例存在色素沉着,2 例有阴 蒂肥大,1 例有阴茎肥大。见表 2。
| 表 2临床表现 [ 例(%)] |
单纯男性化型患儿中各实验室指标水平在治 疗前后差异无统计学意义(P>0.05);失盐型患儿 治疗后睾酮、雌二醇和血钾水平均较治疗前显著 降低(P<0.05),而皮质醇和血钠水平较治疗前明显回升(P<0.05),ACTH 在治疗前后差异无统计 学意义(P>0.05);非典型型患儿仅有皮质醇在 治疗后显著好转(P<0.05)(表 3)。共 41 例患 儿行肾上腺影像学检查,其中单纯男性化型患儿 11 例,阳性 8 例;失盐型患儿 21 例,阳性 13 例; 非典型型患儿 9 例,阳性 4 例。3 例单纯男性化型 及 6 例非典型型患儿行 ACTH 激发试验,结果均 阳性。
| 表 3 两组实验室指标检测结果比较[x±s或 P50(P25, P75)] |

52 例患儿中有 10 例确诊后放弃治疗,其余 42 例均应用激素治疗,治疗过程中自动出院 9 例。 7 例出现肾上腺危象,予 5% 碳酸氢钠、0.09% 氯 化钠静脉滴注纠酸扩容,琥珀酸氢化可的松每日 100~150 mg/m2 静脉滴注,1~3 d 后逐渐减量,1 周 左右改为口服醋酸可的松每日 20~25 mg/m2 。26 例 在应用糖皮质激素基础上加用 9-α 氟氢可的松补充盐皮质激素。12 例患儿住院期间合并有感染表现 (包含肺炎、败血症等)。出院后电话随访 22 例, 其中单纯男性化型 7 例,中位随访时间 17.7 个月, 失盐型 13 例,中位随访时间 35.1 个月,非典型型 2 例,中位随访时间 2.5 个月。所有随访患儿均继 续口服激素,9 例感染后病情加重再次住院,8 例 患儿有性早熟表现,出现明显生长加速及第二性 征发育、骨龄提前,其余生长发育尚正常,感染 后亦无病情加重。 3 讨论
就 CAH 发病男女比例而言,因女性出生时具 有不同程度的男性化症状,往往不易漏诊,而男 性患者、尤其是非失盐型患儿,外生殖器形态多 正常,故不易诊断,据《诸福棠实用儿科学》资 料显示女性患儿发病多于男性,比例约为 2:1。然 而本研究数据则显示男性发病更多,与其他学者 汇报类似 [3, 4],这可能与儿科医生的认知水平提高、 更多男性非典型病例得以早期发现有关。另外, 由本组资料可以看到,52 例患儿起病年龄多在 1 个月以内(41 例),主要表现为呕吐、腹泻、 脱水、电解质紊乱,其中有 7 例出现肾上腺危象、 全部发生于失盐型,提示早期症状识别对诊断本 病的重要性。本病因属于常染色体隐性遗传病, 多有家族史,本研究共识别了 4 例阳性家族史患 者,但因我国多数家庭为独生子女,故常常不能 在第一产时得以明确。基因检测是确诊本病的一 个重要手段,对于治疗及预后的判断亦有价值。 现已发现 90% 以上的 CAH 患儿是由于 21- 羟化酶 基因缺陷所致 [5, 6]。此缺陷基因位于第 6 号染色体 短臂上(6p21. 3),介于 HLA-B 和 HLA-DR,迄 今已发现多种基因突变类型。尽管基因检测具有 诸多优势,然而由于经济、社会观念等原因,其 广泛开展尚有待时日。ACTH 激发对于临床不典型 患儿具有重要价值,本研究对 9 名存疑者进行了 相关试验、结果均阳性,激发试验较为简便易行, 提示其较大的应用价值。
从临床症状来说,单纯男性化型,在女孩早 期表现为假两性畸形,青春期则表现为原发性闭 经伴外生殖器发育异常。本组数据亦表明 13 例女孩全部有阴蒂肥大,且女性发病比例高于男性。 失盐型患儿因可并发危象故临床上常是观察的重 中之重,本型多在小婴儿发病,本研究资料统计 28 人中有 27 例在新生儿期发病,而在 28 例患儿中, 25 例有色素沉着,提示体格检查的重要性,需要 指出的是有相当部分患儿(17/28 人)存在喂养困 难和(或)体重增长缓慢,这类症状在临床工作 中多无特异性,需加以重视。非典型型患儿临床 多无症状,本资料中大部分患儿因外阴色素沉着 和生后 17-α 羟孕酮筛查异常才行就诊确诊。
本研究患儿在确诊 CAH 后均口服氢化可的松 (皮质功能危象者先静脉应用氢化可的松),失 盐型患儿加服食盐及 9-α 氟氢可的松。CAH 治疗 期间难以单凭某一指标作为代谢控制良好与否或 药物剂量调节的可靠依据,临床上可以结合 17-α 羟孕酮、睾酮 ( 女性患儿及青春期前男性患儿 ) 进 行综合分析,有条件时还可测定雄烯二酮 [7]。本研 究显示,在治疗过程中三种类型,尤其是失盐型 患儿,皮质醇、睾酮、雌二醇、电解质水平均有 好转。应当注意的是:ACTH、皮质醇存在明显昼 夜节律性,且个体间差异较大,本文有 7 例皮质 醇增高表现患儿,考虑与抽血检测时间未注意激 素分泌节律有关,结果须慎重看待。
本病的转归在于两个阶段,其一:急性期的 逆转,特别是在失盐型患儿,若有呕吐、腹泻、低钠、 高钾、酸中毒应高度怀疑本病,结合婴幼儿肤色黑、 外生殖器改变,做到早期诊断及治疗并无困难; 其二:长期激素控制与随访,对此的评价主要在 于骨龄(身高)、外生殖器的形态,早期开展激 素替代可明显改善预后 [3],甚至可达到父母遗传身 高 [8],本研究 22 例患儿确诊后即采用长期激素替 代,但有 8 例患儿表现出性早熟,或许与这些患 儿激素替代开始年龄较晚、缺乏规律用药有关, 毕竟外源性的氢化可的松尚不能完全模仿 ACTH 脉冲性分泌与随之而来的可的松分泌脉冲之间密 切时效性关系 [9]。
总之,通过普及新生儿筛查,对相关特异症 状、体征做出早期识别,预防及控制肾上腺危象, 尽早激素替代对于改善患儿预后具有重要意义, 部分患儿可获得正常或接近正常的遗传身高。
| [1] | Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency:an endocrine society clinical practice guideline[J]. J Clin Endocrinol Metab, 2010, 95(9):4133-4160. |
| [2] | 胡亚美, 江载芳. 诸福棠实用儿科学[M]. 第7 版. 北京:人 民卫生出版社, 2011:2022-2024. |
| [3] | Speiser PW, Azziz R, Baskin LS, et al. A summary of the endocrine society clinical practice guidelines on congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency[J]. Int J Pediatr Endocrinol, 2010, 2010:494173. |
| [4] | 宫丽霏, 叶军, 韩连书, 等. 先天性肾上腺皮质增生症早期 治疗回顾分析[J]. 临床儿科杂志, 2013, 31(1):36-39. |
| [5] | 万乃君, 汪伶伶, 王怡萍. 先天性肾上腺皮质增生症/21-羟 化酶缺陷44 例分析[J]. 实用儿科临床杂志, 2011, 26(8):569-571. |
| [6] | 林小梅, 吴本清, 黄进洁, 等. 先天性肾上腺皮质增生症1 例CYP21A2 基因突变分析[J]. 中国当代儿科杂志, 2013, 15(11):942-947. |
| [7] | 梁玲, 辛颖. 47 例先天性肾上腺皮质增生症的临床特点及诊 治分析[J]. 中国优生与遗传杂志, 2014, 1(1):119-121. |
| [8] | 丁翊君, 林影, 王亚娟, 等. 新生儿先天性肾上腺皮质增生 症20 例临床分析[J]. 中国新生儿科杂志, 2010, 4(25):216-218. |
| [9] | 孙文鑫. 先天性肾上腺皮质增生症的长期治疗及疗效评价[J]. 实用儿科临床杂志, 2009, 8(24):569-571. |