 2015, Vol. 17
2015, Vol. 17



2. 福州儿童医院儿科, 福建 福州 350001;
3. 广西桂林医学院第一附属医院儿科, 广西 桂林 541000
噬血细胞综合征(HPS)又称噬血细胞淋巴组织细胞增生症(HLH),是一种由多种致病因素所致淋巴细胞和组织细胞大量增殖、活化伴高细胞因子血症的机体免疫调节异常性疾病[1]。临床特征表现为持续性发热超过1周、不同程度血细胞减少、肝脾和淋巴结肿大、凝血功能障碍、NK细胞活性降低、高甘油三酯血症或低纤维蛋白原血症、骨髓出现噬血现象、中枢神经系统受累等多脏器病变等[2]。HLH分为原发性HLH和获得性HLH,其中原发性HLH包括家族性HLH(FHL)和免疫缺陷综合征相关性HLH。其中FHL主要发生在年龄小于2岁的儿童中,发病年龄越小,病死率越高[3]。
在目前大量国内外关于HLH分子遗传学研究中,FHL已被证实与一些基因外显子编码序列基因突变导致编码蛋白质功能缺陷有关。根据致病基因不同,FHL被分为5个亚型:FHL1(位于 9q21.3-22 的未知基因)、FHL2(PRF1)、FHL3(UNC13D)、FHL4(STX11)及 FHL5(STXBP2),而PRF1基因突变占FHL的20%~50%[3,4,5]。目前已发现70多种PRF1编码序列基因突变与FHL发病有关。目前对PRF1基因研究主要集中在编码蛋白质序列基因突变与FLH发病的关系,关于穿孔素基因多态性位点对FHL发病的易感性相关报道较少。国外相继有文章报道PRF1基因多态性位点A91C可能影响穿孔素蛋白正确折叠,从而导致穿孔素表达,提示PRF1基因多态性位点可能在穿孔素基因转录调控、蛋白质翻译中起重要作用而影响穿孔素正常表达[5,6,7,8]。因此,本研究对48例HLH患儿PRF1基因编码区碱基序列包括3个外显子和2个内含子进行基因多态性筛查,旨在揭示穿孔素基因多态位点在HLH患儿中的发生情况,并探讨PRF1基因多态性是否与儿童HLH发病存在易感相关性。
1 资料与方法 1.1 研究对象选取2009年1月至2013年12月在广西医科大学第一附属医院确诊为HLH患儿48例为研究对象,其中男26例,女22例;发病年龄为6个月至14岁,中位年龄为3岁;诊断标准依据国际组织细胞协会修订的《HLH-2004诊断和治疗指南》[9],均无HLH家族病史。另选取于我院行健康体检的儿童100例作为对照组,其中男56例,女44例;平均年龄3岁9个月。两组间年龄、性别比较差异均无统计学意义(P>0.05),具有可比性。本研究已获得所有入组对象监护人的知情同意。
1.2 主要试剂与仪器血液基因组DNA提取试剂盒、2×Master Mix和1 000 bp DNA Ladder(北京康为世纪生物科技有限公司,中国);引物合成(上海生工生物工程技术服务有限公司,中国);多通道 PCR 扩增仪(PTC-220,美国);恒温水浴箱(DK-420,中国);电泳仪(DYY-86 型,中国);Gel Doc EQ凝胶成像系统(Bio-RAD,美国)。
1.3 DNA提取分别采集HLH组患儿及对照组儿童外周静脉血2 mL置于EDTA抗凝管中;应用血液基因组DNA提取试剂盒从两组外周血标本中提取DNA。
1.4 引物设计应用 Primer Premier 5.0及NCBI primer designing tool对人 PRF1 基因外显子和内含子序列(GenBank 序列号:NG_009615)进行引物设计,具体引物序列见表 1。
| 表 1 扩增穿孔素基因外显子及内含子的引物序列 |
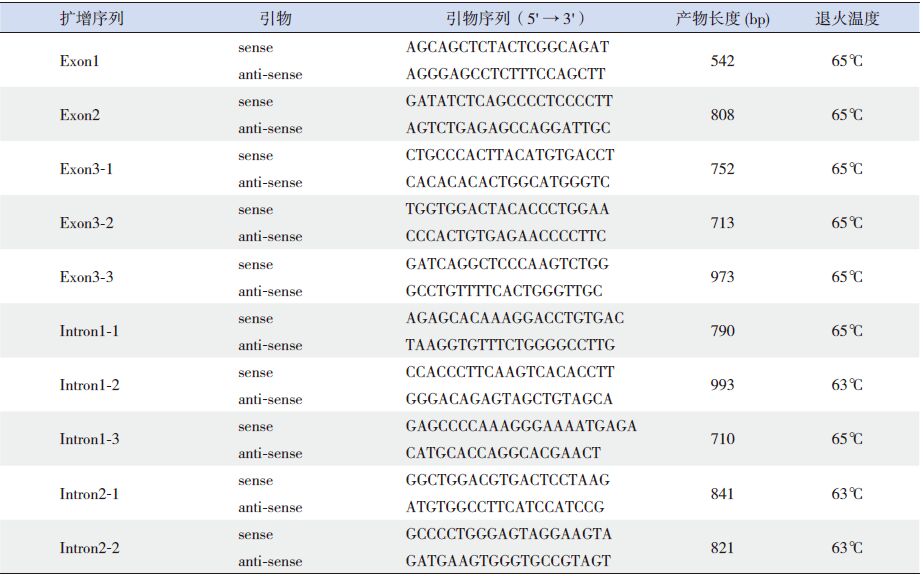
PCR反应体系为50 μL:2×Master Mix 25 μL、10 μmol/L上下游引物各2 μL、DNA模板2 μL,ddH2O 19 μL。PCR反应条件:94℃预变性 4 min;94℃变性45 s,退火(温度见表 1)45 s,72℃延伸1 min,共 35个循环;72℃再延伸 7 min。取 5 μL PCR产物在2% 琼脂糖凝胶中进行电泳(150 V,25 min),以 1000 bp DNA Ladder 为相对分子质量标准。将凝胶置于 Gel成像系统进行成像并分析。PCR产物测序及序列比对:PCR产物交由上海生工生物工程技术服务有限公司进行测序。测序结果采用美国国家生物技术信息中心(NCBI)Genebank BLAST软件进行在线比对。
1.6 统计学分析采用 SPSS 16.0 统计软件对数据进行统计学分析,计数资料采用百分率(%)表示,两组间比较采用χ2检验,若1≤理论频数(T)<5时,采用连续校正χ2检验,若T<1时,采用Fisher确切概率法,P<0.05为差异有统计学意义。应用SHEsis软件进行连锁不平衡分析(frequency<0.01则被忽略)。
2 结果 2.1 HLH患儿临床特征分析48例HLH患儿中,持续发热超过1周48例(100%);肝肿大48例(100%);脾肿大45例(94%);血细胞三系减少15例(31%),两系减少16例(33%);纤维蛋白原≤1.5 g/L 27例(56%);甘油三酯≥3.0 mmol/L 36例(75%);NK细胞比值低于6% 41例(85%);铁蛋白≥500 ng/L 45例(94%);骨髓出现噬血现象36例(71%)。
2.2 PRF1基因多态性位点筛查结果48例HLH患儿中,在PRF1基因编码序列中共发现3个SNP位点:其中位于外显子2上的R4C(rs12161733)2例(4%);外显子3上的g.8877C>T(rs885821)14例(29%),g.8955C>T(rs885822)44例(92%)。而在非编码序列中共发现7个SNP位点:其中位于内含子1上的g.5179delG(rs55974750)25例(52%),g.5452C>T (rs3758562)46例(96%),g.5936T>C(rs6480460)47例(98%);内含子2上的g.7745T>C(rs80019657)10例(21%);3'非翻译区上的g.9819G>A (rs1889490)41例(85%),g.9812C>T(rs6480459)2例(4%),g.10164G>A(rs375066000)2例(4%)。以上10个SNP位点在HLH组和对照组中的基因型及等位基因频率分布差异均无统计学意义(P>0.05),见表 2。
| 表 2 PRF1 基因各多态性位点的基因型和等位基因频率在两组中的分布比较 [例(%)] |
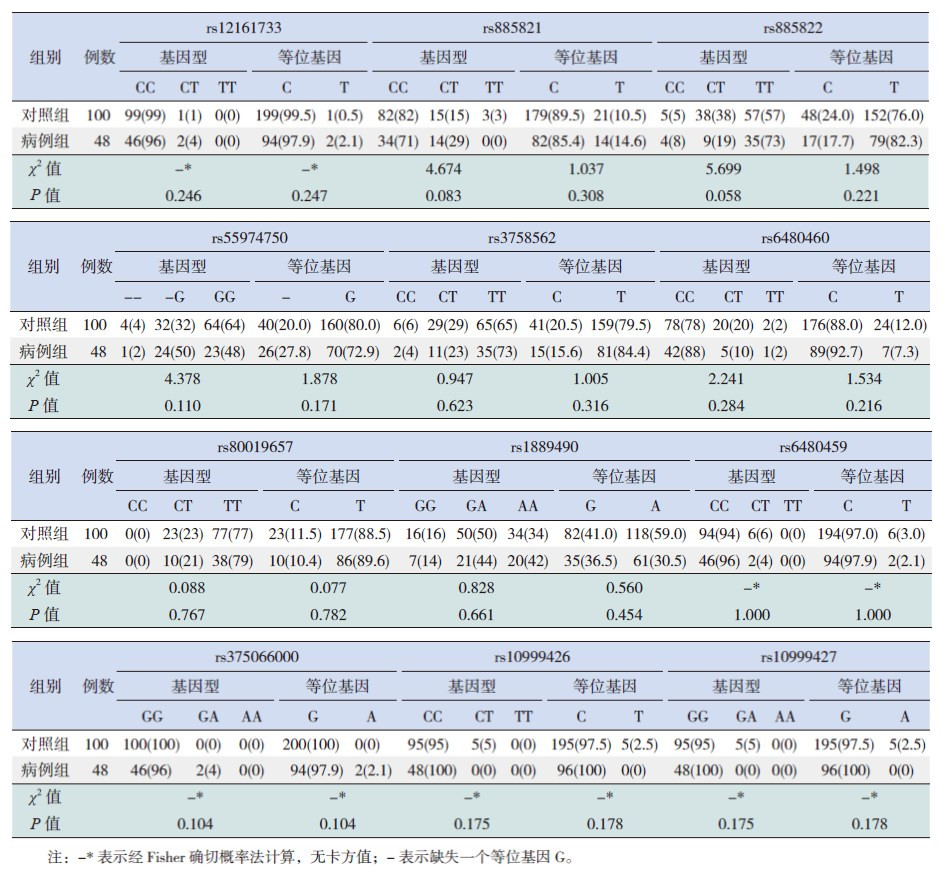
在PRF1基因第2内含子上发现另外2个SNP位点:g.7494C>T(rs10999426)和g.7473G>A(rs10999427),分别仅在5例对照组儿童中发现,以上SNP位点在两组中的基因型及等位基因频率分布差异无统计学意义(P>0.05),见表 2。
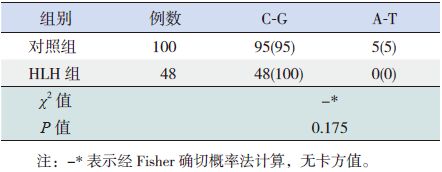
连锁不平衡分析提示rs10999426和rs10999427紧密连锁(D=1,r2=1),但他们构建的单体型在HLH组和对照组中的分布频率差异无统计学意义(P>0.05),见表 3。
| 表 3 两组SNP 位点单体型分布 [例(%)] |
人类穿孔素基因(PRF1)定位于10q21-22,包括3个外显子和2个内含子,编码蛋白质的1 668个碱基仅位于外显子2和外显子3,共编码555个氨基酸[10,11]。穿孔素基因编码穿孔素蛋白前体,包括21个氨基酸组成的信号肽、膜攻击复合体/穿孔素结构域(membrane-attack complex,MAC/Perforin domain)、表皮生长因子样结构域(EGF-LIKE domain)和蛋白激酶C保守区2[11,12]。穿孔素主要储存在细胞毒性T淋巴细胞(CTL)和NK细胞内囊泡中,是参与细胞毒性细胞杀伤靶细胞的一个重要分子,在免疫监视、免疫调控中起着重要作用,其功能异常可导致多种疾病。国内外大量分子遗传学研究提示穿孔素基因变异可能与多种疾病相关,包括I型糖尿病、多发性硬化、恶性血液病如急性白血病、淋巴瘤、HLH等有关[13,14,15,16,17,18]。
1999年Stepp等[18]首次证实了PRF1与FHL2有关。国内外大量研究表明,FHL患者缺乏诱导细胞凋亡和细胞毒杀伤细胞活性的能力,与“穿孔素/颗粒酶-细胞死亡途径”有关。PRF1基因突变可导致穿孔素在CTL、NK细胞表面表达减少甚至不表达,从而不能锚定靶细胞启动细胞凋亡机制,导致颗粒酶无法进入靶细胞诱导靶细胞凋亡,而致使淋巴细胞和组织细胞的大量增值,分泌大量细胞因子,导致FHL2发病[19]。不同国家PRF1基因突变特点不同,如50delT(L17fsX22)与美 洲人有关[20],在日本,1090-91delCT(L364fsX)是最常见的基因突变位点,207delC则为第二常见的基因位点[21,22];在土耳其,最常见的基因突变位点是1122 G>A(W374X)[23];国内有研究报道了S168N、T450M较为常见,rs885821、rs885822为最常见的SNP位点[24,25,26],但关于PRF1是否存在基因多态性及与HLH存在易感相关性尚无相关报道。
本研究在PRF1基因编码序列中共发现3个SNP。其中R4C(rs12161733)仅在2例患儿(4%)和1例正常儿童(1%)中出现,发生率低,其在病例组和对照组的基因型和等位基因频率差异无统计学意义,提示该单核苷酸变异导致的氨基酸替换可能并不影响信号肽结构改变,与FHL发病易感相关性不大。而g.8877C>T(rs885821)、g.8955C>T(rs885822)均未导致氨基酸改变,不影响穿孔素蛋白编码,且在病例组及对照组中均广泛存在,为常见的基因多态性位点,故对FHL发病无意义。
在非编码序列中共发现9个SNP位点。其中rs55974750、rs3758562、rs6480460、rs80019657、rs1889490在HLH组的分布频率分别为52%、96%、98%、21%、85%,而在对照组中的分布频率分别为36%、94%、98%、23%、84%,其等位基因频率在HLH组和对照组间的差异均无统计学意义。rs6480459仅在2例HLH患儿(4%)和6例正常儿童(6%)中发现,而rs375066000仅在2例HLH患儿(4%)中发现,均为杂合子,发生率低,其基因型和等位基因频率在HLH组及对照组中的差异亦无统计学意义。本研究提示以上7个SNP位点在穿孔素基因转录、蛋白合成过程中参与调控作用的可能性不大,对FHL发病无明显易感性。
此外,连锁不平衡分析提示rs10999426 和 rs10999427紧密连锁,此两位点构建的A-T单体型在HLH组和对照组中的分布频率分别为0%和5%,但A-T单体型在两组间的分布差异无统计学意义。因此,A-T单体型与FHL无明显易感相关性。该单体型虽仅在对照组中发现,但发生率低,可能并不是FHL的保护性因素。
FHL起病急,发展快,病死率很高,预后差,病情易反复。国内外研究已经发现了影响该病发生发展及预后的诸多因素[27],其中穿孔素基因异常是重要的因素之一。本研究中提示穿孔素基因多态性与HLH易感性可能性不大,但值得说明的是,本研究中48例HLH患儿穿孔素基因编码序列未发现基因突变,如错义突变、移码突变等,仅对穿孔素基因外显子及内含子区域进行基因多态性筛查,且并未对其他FHL相关基因如UNC13D、STX11、STXBP2等进行基因分析。而穿孔素基因增强子、启动子区域等其他区域序列及其他相关基因是否存在碱基变异在基因调控中起作用,尚有待进一步研究探讨,以期发现能够揭示对儿童 HLH病因、临床诊治及预后有意义的分子生物学标志。
| [1] | Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts[J]. Curr Opin Pediatr, 2012, 24(1): 9-15. |
| [2] | Janka GE. Fami l ial and acquired hemophagocytic lymphohistiocytosis[J]. Annu Rev Med, 2012, 63: 233-246. |
| [3] | Gholam C, Grigoriadou S, Gilmour KC, et al. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management[J]. Clin Exp Immunol, 2011, 163(3): 271-283. |
| [4] | Ta n g YM, Xu XJ. Advances in hemophagocytic lymphohistiocytosis: pathogenesis, early diagnosis/differential diagnosis, and treatment[J]. Scientific World Journal, 2011, 11(1): 697-708. |
| [5] | Voskoboinik I, Thia MC, Trapani JA. A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis[J]. Blood, 2005, 105(12): 4700-4706. |
| [6] | Sanchez IP, Leal-Esteban LC, Alvarez-Alvarez JA, et al. Analyses of the PRF1 gene in individuals with hemophagocytic lymphohystiocytosis reveal the common haplotype R54C/A91V in Colombian unrelated families associated with late onset disease[J]. J Clin Immunol, 2012, 32(4): 670-680. |
| [7] | Martinez-Pomar N, Lanio N, Romo N, et al. Functional impact of A91V mutation of the PRF1 perforin gene[J]. Hum Immunol, 2013, 74(1): 14-17. |
| [8] | Trambas C, Gallo F, Pende D, et al. A single amino acid change, A91V, leads to conformational changes that can impair processing to the active form of perforin[J]. Blood, 2005, 106(3): 932-937. |
| [9] | H e n t e r J I, H o r n e A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis[J]. Pediatr Blood Cancer, 2007, 48(2): 124-131. |
| [10] | Zur Stadt U, Beutel K, Kolberg S, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A[J]. Hum Mutat, 2006, 27(1): 62-68. |
| [11] | Lichtenheld MG, Olsen KJ, Lu P, et al. Structure and function of human perforin[J]. Nature, 1988, 335(6189): 448-451. |
| [12] | An O, Gursoy A, Gurgey A, et al. Structural and functional analysis of perforin mutations in association with clinical data of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) patients[J]. Protein Sci, 2013, 22(6): 823-839. |
| [13] | Kägi D, Odermatt B, Seiler P, et al. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice[J]. J Exp Med, 1997, 186(7): 989-997. |
| [14] | Cappellano G, Orilieri E, Comi C, et al. Variations of the perforin gene in patients with multiple sclerosis[J]. Genes Immun, 2008, 9(5): 438-444. |
| [15] | Manso R, Rodriguez-Pinilla SM, Lombardia L, et al. An A91V SNP in the perforin gene is frequently found in NK/T-Cell lymphomas[J]. PLoS One, 2014, 9(3): e91521. |
| [16] | Yang L, Liu H, Zhao J, et al. Mutations of perforin gene in Chinese patients with acute lymphoblastic leukemia[J]. Leuk Res, 2011, 35(2): 196-199. |
| [17] | Trapani JA, Thia KYT, Andrews M, et al. Human perforin mutations and susceptibility to multiple primary cancers[J]. Oncoimmunology, 2013, 2(4): e24185. |
| [18] | Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis[J]. Science, 1999, 286(2): 1957-1959. |
| [19] | Arico M, Nespoli L, Maccario R, et al. Natural cytotoxicity impairment in familial haemophagocytic lymphohistiocytosis[J]. Arch Dis Child, 1988, 63(3): 292-296. |
| [20] | Lee SM, Villanueva J, Sumegi J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis[J]. J Med Genet, 2004, 41(2): 137-144. |
| [21] | Ishii E, Ohga S, Imashuku S, et al. Review of hemophagocytic lymphohistiocytosis (HLH) in children with focus on Japanese experiences[J]. Crit Rev Oncol Hematol, 2005, 53(3): 209-223. |
| [22] | Nagai K, Yamamoto K, Fujiwara H, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes[J]. PLoS One, 2010, 5(11): e14173. |
| [23] | Okur H, Balta G, Akarsu N, et al. Clinical and molecular aspects of Turkish familial hemophagocytic lymphohistiocytosis patients with perforin mutations[J]. Leuk Res, 2008, 32(6): 972-975. |
| [24] | Lu G, Xie Z, Shen K, et al. Mutations in the perforin gene in children with hemophagocytic lymphohistiocytosis[J]. Chin Med J (Engl), 2009, 31(23): 2851. |
| [25] | 谢淑佩, 徐忠金, 范小菊. 噬血细胞综合征患儿中穿孔素基因突变的研究[J]. 江西医药, 2013, 48(9): 810-811. |
| [26] | 刘红星, 童春容, 王卉, 等. 家族性噬血细胞性淋巴组织细胞增多症一例病因和遗传学研究[J]. 中华内科杂志, 2011, 50(2): 132-135. |
| [27] | 陆文娴, 罗建明. 儿童噬血细胞性淋巴组织细胞增生症的预后因素分析[J]. 中国当代儿科杂志, 2012, 14(8): 593-597. |