 2015, Vol. 17
2015, Vol. 17



甲基丙二酸血症(methylmalonic academia,MMA)合并高同型半胱氨酸血症(简称合并型MMA)是MMA较常见的类型[1],也是我国MMA患者最常见的生化表型,包括cblC、cblD和cblF3 种亚型,其中cblC型是我国合并型MMA的主要病因[2]。合并型MMA患者的临床表型远较单纯MMA更为复杂,从严重的早发型多系统受累到轻型晚发型,个体差异很大。目前国内关于cblC型MMA的研究多为临床及基因诊断报道,本研究在此基础上对16例cblC型MMA患者进行了基因型与临床表型及疗效之间关系的探讨。 1 资料与方法 1.1 研究对象
2004年12月至2014年12月在我院发育儿科就诊的合并型MMA患儿16例,其中男12例,女4例,发病年龄中位数为4.5个月(范围:生后5d~9岁10个月)。1例来自新生儿遗传代谢病筛查,其他15例主要以营养不良、贫血伴或不伴全血细胞减少、抽搐、嗜睡、生长发育落后和(或)倒退及鱼鳞病就诊,父母均非近亲婚配。所有检查均获得患儿家长的知情同意。 1.2 方法
(1)合并型MMA的诊断标准:符合MMA的临床表现,经尿气相色谱/质谱(gaschromatography mass spectrometry,GS/MS)技术检测尿液中甲基丙二酸、血串联质谱(tandem massspectrometry,MS/MS)技术检测血中丙酰肉碱(C3)与乙酰肉碱(C2)的比值(C3/C2)同时增高并排除维生素B12缺乏等继发因素伴血清同型半胱氨酸升高,可合并甲硫氨酸(methionine,Met)降低的患儿诊断为合并型MMA[3]。
(2)MMA的分型:按发病年龄分为早发型(发病年龄≤1岁)及晚发型(发病年龄>1岁)[4-5]。本研究又按临床表型分为轻型、中间型及重型。晚发型患儿均为轻型;早发型患儿包括中间型及重型,其中生后1个月内发病者为重型,生后1个月至1岁发病同时出现下列临床表现中的1种及以上时为重型:死亡、代谢危象、严重的发育迟缓、癫癎、危及生命的全血细胞减少及小头畸形,否则为中间型。
(3)辅助检查:取患儿及其父母外周静脉血,进行MMACHC基因检测,该基因为CblC型合并型MMA的编码基因。同时完善患儿血、尿常规、血气分析、血氨、血糖、血乳酸、肝肾功能和心肌酶谱测定、脑CT 或MRI 扫描、脑电图、智力测定等辅助检查。 2 结果 2.1 cblC型MMA患儿临床资料
16例cblC型MMA患儿中15例为早发型,1例为晚发型(轻型)。15例早发型又包括4例中间型和11例重型。死亡3例,代谢危象3例,抽搐7例,小头畸形7例,发育迟缓10例,眼部问题6例。16例患儿均行头部MRI检查,4例无异常(重型及轻型各1例,中间型2例),12例异常(重型10例,中间型2例),表现为脑外间隙增宽或合并脑室增大9例,脑白质异常5例,胼胝体发育不良4例,脑积水5例,蛛网膜下腔出血1例。9例患儿行脑电图检查,1例无异常(中间型),1例广泛电压抑制(重型),7例抽搐患儿脑电图检查均异常,表现为广泛电压抑制1例,高度失节律1例,局灶性或多灶性癎样放电5例。急性发作期患儿均有不同程度的代谢性酸中毒(pH7.05~7.28),部分患儿乳酸及血氨升高。血液系统异常11例,其中全血细胞减少4例(重型),肝功能异常3例(重型)。16例患儿基因型与表型特点见表 1。
| 表 1 16例合并型MMA患儿临床资料 |

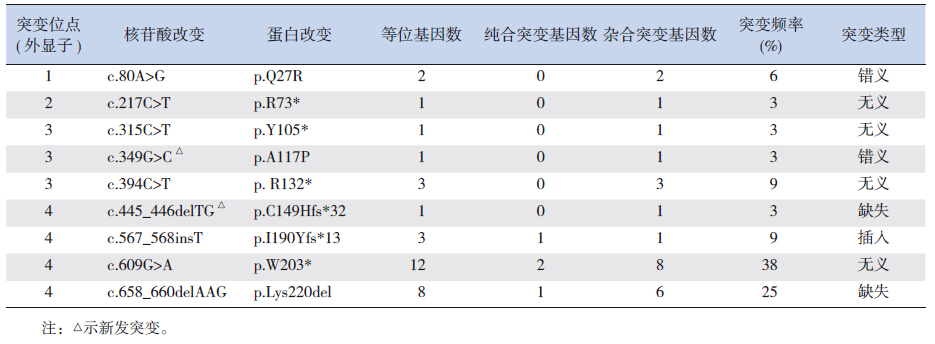
本组患儿中共检出9种MMACHC 基因突变(表 2)、4种突变类型(无义、插人、缺失及错义突变),以无义突变为主(4/9,44%)。c.609G>A(2例纯合突变、8例杂合突变)和c.658_660delAAG突变(1例纯合突变、6例杂合突变)为最常见的早发型突变(87%,13/15),前者突变频率最高(38%,12/32),后者次之(25%,8/32)。这2个突变导致的杂合突变是最常见的复合杂合突变(4/12,33%),也是存活患儿中最常见、表型最重的突变形式(4/13,31%),受累患儿以小头畸形、癫癎及严重的发育迟缓为主要表现。c.609G>A突变是最常见的纯合突变(2/4,50%),临床表型为中间型。c.658_660delAAG纯合突变为首次报道,患儿表型重。2例c.567_568insT突变(1例纯合突变,另1例c.567_568insT/c.658_660delAAG杂合突变)患儿均死亡。3例c.394C>T 突变均为早发型复合杂合突变(2例中间型、1例重型)。2例c.80A>G突变也均为复合杂合突变(1例轻型,1例死亡)。c.445_446delTG及c.349G>C 突变国内外尚未见报道,突变频率均为3%(1/32);50例健康人中未检测到此两种突变。2例新发突变的基因测序结果见图 1。
| 表 2 16例患儿MMACHC基因突变检测结果分析 |
 |
图 1 2例cblC型MMA患儿新发突变基因DNA测序图 A:患儿男,37 d,MMACHC基因exon4缺失突变;B:患儿男,9 岁10 个月,MMACHC基因exon3错义突变。箭头所示为基因突变位点。 |
分析临床资料、营养发育状况、合并症等情况后,对患儿进行相应的个体化饮食与药物治疗,针对惊厥、腹泻、感染、营养不良等合并症进行对症干预。给予羟钴胺或氰钴胺/维生素B12(1 mg/周~1 mg/d)肌肉注射,口服叶酸或亚叶酸钙(10~30 mg/d)、左卡尼汀[30~200 mg/(kg · d)]、甜菜碱(0.5~3g/d)及维生素B6(10~30mg/d),必要时给予蛋氨酸(200mg/d);正常饮食。其中维生素B12/氰钴胺治疗8例,羟钴胺治疗3 例。最常见、表型最重的3例复合杂合突变(c.609G>A/c.658_660lAAG)患儿经治疗小头畸形、眼科问题均无明显改善,但2例由维生素B12/氰钴胺换为羟钴胺后抽搐减轻,另1例全疗程羟钴胺治疗后无抽搐出现。c.658_660delAAG纯合突变为首次报道,患儿表型重、疗效差,由维生素B12/氰钴胺换为羟钴胺后抽搐缓解。c.394C>T杂合突变中2例中间型患儿(c.394C>T/c.609G>A及c.394C>T/c.658_660delAAG)维生素B12/氰钴胺疗效较好,另1例重型患儿(394C>T/217C>T)表型严重,维生素B12/氰钴胺治疗后抽搐缓解但遗留有严重的发育迟缓。c.80A>G杂合突变2例,其中1例轻型患儿经羟钴胺治疗临床表型完全正常,智力商数为105,另1例死亡。所有患儿经过治疗后临床症状及生化表现明显改善,尿中甲基丙二酸浓度显著下降。除1 例晚发型患儿外,其他患儿治疗后均有不同程度的智力和运动发育落后。 3 讨论
合并型MMA是MMA中最常见的类型,也是我国MMA的主要类型[6],为钴胺素代谢异常所致腺苷钴胺素和甲基钴胺素合成缺陷,线粒体内甲基丙二酰辅酶A 变位酶及胞浆内甲硫氨酸合成酶的活性下降,最终导致血清/ 尿甲基丙二酸、血清同型半胱氨酸增高及血清甲硫氨酸减少。合并型MMA 分为cblC、cblD及cblF 3型,其中cblC型最为常见,其编码基因为MMACHC(OMIM277400),该基因定位于常染色体1p34,包含4个编码外显子及1 个非编码外显子[4]。cblC蛋白含282个氨基酸,有两个功能结构域:经典的钴胺素结合区(122-HXXGX126-154GG-156)及与TonB 折叠结构功能相似的C-末端(残基185-282区,C-末端可能参与钴胺素转运所需的质子泵能量转化,以形成钴胺素代谢所必须的构象)[1]。cblC 蛋白主要存在胞浆内,其主要功能是催化CNCbl还原脱氰和合成腺苷钻胺素及甲基钴胺素,当MMACHC基因发生突变时,机体内甲基丙二酸及同型半胱氨酸蓄积伴甲硫氨酸降低。
目前国内外已报道MMACHC基因突变近80种,国内30种[2,7-10]。我国cblC缺陷型MMA的基因型与欧美国家显著不同。欧美国家突变频率最高的是c.271dupA(R91Kfs*14)(40%~55%)[4-5,11-12],c.271dupA纯合突变几乎均为早发型,而我国最常见的早发型突变是c.609G>A(48.1~67.8%)及c.658_660delAAG(7.1%~13.9%)[2,6-10]。基因突变的人群差异可能与方舟效应及遗传漂变有关[2]。与文献报道类似[2,4,9-10],c.609G>A及c.658_660delAAG是本研究病例最常见的早发型突变,进一步证明了有关这两个突变是我国早发型cblC型MMA热点突变的论点[9-10]。本研究发现二者导致的复合杂合突变(c.609G>A/c.658_660delAAG)是存活患儿中最常见、表型最重、预后最差的突变,受累患儿小头畸形、眼部问题治疗后并无明显改善,但羟钴胺有助于抽搐控制。c.609G>A为无义突变可造成第203位色氨酸翻译提前终止,c.658_660delAAG为框移突变,导致蛋白质翻译的第220位赖氨酸缺失,这两个突变均产生了截短蛋白从而造成MMACHC蛋白功能缺陷。文献报道中c.609G>A纯合突变及c.609G>A/c.658_660delAAG复合杂合突变多为早发型[2, 7, 13],仅各见1例晚发型病例[7]。本研究发现609G>A纯合突变患儿临床表型较轻(中间型),而c.658_660delAAG纯合突变临床表型重、疗效差;c.567_568insT突变造成从第190位异亮氨酸开始发生移码突变,以往临床资料多为与c.609G>A导致的早发型杂合突变,其纯合突变及与c.658_660delAAG的杂合突变为本文首次报道,2例患儿均死亡。结合文献推测c.567_568insT突变主要见于我国早发型患儿,表型重、预后差,其突变变基因型与表现型之间的关系有待进一步确定。MMACHC蛋白结构分析提示c.609G>A、c.658_660delAAG及c.567_568insT突变位于C-末端结构区中,均可能影响钴胺素转运所需的质子泵能量转化[7],c.658_660delAAG突变也位于同源序列的高度保守区,提示该位置对蛋白功能影响较大[9]。本研究中c.394C>T和c.80A>G突变分别与其类型突变导致的复合杂合突变临床表型及疗效均有很大差异。2例中间型c.394C>T杂合突变患儿疗效较好。另1例重型患儿表型严重,治疗效果差。c.394C>T突变在印度、巴基斯坦、沙特阿拉伯、阿富汗、葡萄牙、意大利及我国均有报道[2, 6, 11]。以往认为c.394C>T纯合突变多为晚发型,临床表现较轻、疗效较好[11, 13]。最近有文献指出c.394C>T纯合突变与晚发型并无关联[12, 14]。c.80A>G杂合突变中1例患儿(c.349G>C/c.80A>G杂合突变)是本研究中唯一的轻型晚发型患儿,而另一例患儿(c.80A>G/c.609G>A杂合突变)则死亡。c.349G>C错义突变为新发突变,该突变可使第117位丙氨酸突变脯氨酸,分析该突变临床表型较轻。以往报道的c.80A>G纯合突变及c.80A>G/c.609G>A杂合突变均为早发型[2, 7, 13]。Morel等[11]等认为RNA稳定性或蛋白质的残余功能导致了临床表型不同。基因型与表型关系的复杂性可能与第二个突变的显性负效应、遗传背景、环境作用及饮食习惯等因素相关。本研究中另一种新发突变(c.445_446delTG)位于钴胺素结合区,突变造成从第149位半胱氨酸开始发生移码,在180位密码子处提前终止,产生截短蛋白,从而使MMACH 蛋白丧失活性;这2种新发突变尚需通过体外表达来进一步证实。总之,c.609G>A、c.658_660delAAG及c.567_568insT突变为我国常见的早发型突变、表型严重、疗效较差,c.394C>T及c.80A>G 突变表型差异较大。
由于合并型MMA兼有甲基丙二酸及同型胱氨酸的毒性作用,部分还伴有低甲硫氨酸血症,患者临床表现复杂多样、缺乏特异性,急性发病相对少见,多以慢性进行性神经系统损害为主,相对特异性的症状为包括脑水肿及溶血尿毒综合征在内的弥漫性微血管损伤[12]。本研究中1例患儿(病例10)合并严重的脑积水,最终采取了脑室腹腔分流术治疗,国内有以脑积水起病的cblC型MMA病例报道[15]。低甲硫氨酸血症导致的髓鞘化不良可能是cblC型患者产前脑损伤的主要原因[16]。同型半胱氨酸蓄积可导致血管内皮损伤及N-甲基-D-天冬氨酸受体过度激活而致神经毒性作用[17],该受体激活与惊厥发生有关[18]。溶血尿毒综合征被认为是低甲硫氨酸及高同型半胱氨酸血症共同导致的结果[19]。本研究病例仅2例为急性发病,其他均为慢性进行性神经系统损害为主,未发现溶血尿毒综合征。
目前普遍认为氰钴胺对cblC缺陷患儿疗效较羟钴胺差,这是因为突变的MMACH蛋白对氰钴胺及羟钴胺的结合能力存在内在差异,且与钴胺素的代谢旁路无关[20]。除此之外,本研究还发现疗效与基因型有关,表型越重疗效越差,我国常见的热点突变c.609G>A与c.658_660delAAG表型重、疗效差。虽然16例cblC缺陷患儿经治疗临床症状及实验室指标均明显改善但发育迟缓、生长迟缓、小头畸形、斜视等神经科及眼科的问题并没有得到改善。文献报道羟钴胺通过个体化剂量滴定及缩短用药间期可以达到满意的代谢控制从而使有严重神经损伤的患儿受益[21]。但最近有研究提出cblC 型MMA患者体内存在谷胱甘肽代谢紊乱,过多羟钴胺会干扰谷胱甘肽静态平衡导致氧化型谷胱甘肽增加,还原型谷胱甘肽减少[22]。谷胱甘肽代谢紊乱及其他不明原因的发病机制可能是目前cblC型MMA疗效不满意的原因之一。合并型MMA患儿一般应正常饮食以保证天然蛋白质摄人,从而有利于疾病控制与生长发育。本研究中1例患儿本身存在营养不良及低甲硫氨酸血症,在严格限制蛋白及脂肪摄入后出现皮肤黏膜损害及肛瘘最终死亡,分析可能与过度限制天然蛋白导致医源性低甲硫氨酸血症及继发性肉碱缺乏有关。
总之,我国cblC缺陷型MMA以早发型为主,存在热点突变c.609G>A和c.658_660delAAG,二者导致的复合杂合突变是最常见的突变形式,其临床表型严重、疗效差。c.609G>A纯合突变多见、临床表型更轻、疗效也更好。c.394C>T突变临床表型和疗效差异均较大。与氰钴胺相比羟钴胺对非神经病学表现及异常血尿生化改善更明显,但多数患者仍有神经及眼科学方面问题。另外,本研究发现2个国内外未报道的新突变(c.349G>C和c.445_446delTG)。
| [1] | Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias[J]. J Inherit Metab Dis, 2008, 31(3): 350-360. |
| [2] | Liu MY, Yang YL, Chang YC, et al. Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria[J]. J Hum Genet, 2010, 55(9): 621-626. |
| [3] | 毋盛楠, 韩连书, 叶军, 等. 甲基丙二酸血症患者血串联质谱及尿气相色谱质谱检测分析[J]. 中华医学杂志, 2013, 93(8): 561-565. |
| [4] | Lerner-Ellis JP, Tirone JC, Pawelek PD, et al. Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type[J]. Nat Genet, 2006, 38(1): 93-100. |
| [5] | Fischer S, Huemer M, Baumgartner M, et al. Clinical presentation and outcome in a series of 88 patients with the cblC defect[J]. J Inherit Metab Dis, 2014, 37(5): 831-840. |
| [6] | 张尧, 宋金青, 刘平, 等. 甲基丙二酸尿症伴同型半胱氨酸血症57例临床分析[J]. 中华儿科杂志, 2007, 45(7): 513-517. |
| [7] | Wang F, Han LH, Yang YL, et al. Clinical, biochemical, and molecular analysis of combined methylmalonic acidemia and hyperhomocysteinemia (cblC type) in China[J]. J Inherit Metab Dis, 2010, 33 (Suppl 3): S435-S442. |
| [8] | 刘玉鹏, 马艳艳, 吴桐, 等. 早发型甲基丙二酸尿症160例新生儿期异常表现[J]. 中华儿科杂志, 2012, 50(6): 410-414. |
| [9] | 王斐, 韩连书, 胡宇慧, 等. 甲基丙二酸血症伴同型半胱氨酸血症患儿基因突变分析[J]. 中华儿科杂志, 2009, 47(3), 189-193. |
| [10] | 韩连书, 王斐, 胡宇慧, 等. 甲基丙二酸血症伴同型半胱氨酸血症患儿临床及基因突变分析[J]. 中华内分泌杂志, 2009, 25(8): 405-407. |
| [11] | Morel CF, Lerner-Ellis JP, Rosenblatt DS, et al. Combined methylm alonic aciduria and homocystinuria (cblC): phenotype-genotype correla tions and ethnic-specific observations[J]. Mol Genet Metab, 2006, 88(4): 315-321. |
| [12] | Fischer S, Huemer M, Baumgartner M, et al. Clinical presentation and outcome in a series of 88 patients with the cblC defect[J]. J Inherit Metab Dis, 2014, 37(5): 831-840. |
| [13] | Martina Huemer, Sabine Scholl-Bürgi, et al. Three new cases of late-onset cblC defect and review of the literature illustrating when to consider inborn errors of metabolism beyond infancy[J]. Orphanet J Rare Diseases, 2014, 9: 161. |
| [14] | Debray FG, Boulanger Y, Khiat A, et al. Reduced brain choline in homocystinuria due to remethylation defects[J]. Neurology, 2008, 71(1): 44-49. |
| [15] | 刘黎黎, 侯新琳, 周丛乐, 等. 以脑积水起病的甲基丙二酸尿症合并同型半胱氨酸血症1例报道[J]. 中国当代儿科杂志, 2013, 15(4): 313-315. |
| [16] | Bodamer OA, Sahoo T, Beaudet AL, et al. Creatine metabolism in combined methylmalonic aciduria and homocystinuria[J]. Ann Neurol, 2005, 57(4): 557-560. |
| [17] | Roehrs C, Garrido-Sanabria ER, Silive AC, et a1. Succinate in creases neuronal post-synaptic excitatory potentials in vitro an d induces convulsive behavior through N-methyl-D-aspartated mediated mechanism[J].Neuroscience, 2004, 125(4): 965-971. |
| [18] | Malfatti CR, Perry ML, Schweigert D, et al. Convulsions in-duced by methylmalonic acid are associated with glutamic acid deearboxylase inhibition in rats: a role for GABA in the seizuyes presented by methylmalonic acidemic patients?[J]. Neuroscience, 2007, 146(4): 1879-1887. |
| [19] | Martinelli D, Deodato F, Dionisi-Vici C. Cobalamin C defect: natural history, pathophysiology, and treatment[J]. J Inherit Metab Dis, 2011, 34(1): 127-135. |
| [20] | Froese DS, Zhang J, Healy S, et al. Mechanism of vitamin B12-responsiveness in cblC methylmalonic aciduria with homocystinuria[J]. Mol Genet Metab, 2009, 98(4): 338-343. |
| [21] | Matos IV, Castejón E, Meavilla S. Clinical and biochemical outcome after hydroxocobalamin dose escalation in a series of patients with cobalamin C deficiency[J]. Mol Genet Metab, 2013, 109(4): 360-365. |
| [22] | Pastore A, Martinelli D, Piemonte F. Glutathione metabolism in cobalamin deficiency type C (cblC)[J]. J Inherit Metab Dis, 2014, 37(1): 125-1299. |