 2015, Vol. 17
2015, Vol. 17



Ph染色体阳性急性淋巴细胞白血病(Philadelphia chromosome-positive acute lymphoblastic leukemia,Ph+ ALL)在儿童白血病中占5%左右,具有起病时年龄较大、白细胞高、预后差等独特的临床特征,化疗后的总体生存率仅为20%~30%[1, 2]。随着络氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)的出现,其临床疗效得到明显改善。在COG-AALL0031方案中以强化疗联合伊马替尼治疗儿童Ph+ ALL,完全缓解(complete remission,CR)率达95%,诱导相关病死率为0,5年无病生存率(disease-free survival,DFS)可达(70±12)%,而血缘相关供者造血干细胞移植(allogeneic hematopoietic stem-cell transplantation,allo-HSCT)组DFS则为(65±11)%,无关供者allo-HSCT 组DFS为(59±15)%[3, 4, 5]。尽管上述临床研究具有随访期较短及病例数较少等方面的局限,但靶向治疗的优越性显而易见。因此,allo-HSCT是否作为儿童Ph+ALL治疗的首选成为焦点,同时也引发了化疗联合TKI是否可能替代造血干细胞移植成为部分儿童Ph+ALL治疗一线选择的思考。我院自2008年10月起采用CCLG-ALL2008方案或联合TKI治疗儿童Ph+ ALL,现对其疗效及安全性进行总结分析。 1 资料与方法 1.1 研究对象
研究对象为2008年10月至2013年12月就诊于我院的初诊年龄<15岁的53例Ph+ALL患儿,其中男38例,女15例,平均年龄8±3岁(范围2~15岁),10岁以上患儿占40%。 1.2 诊断标准
参照2014儿童急性淋巴细胞白血病诊疗建议(第四次修订)[6]进行诊断,免疫分型采用多参数流式细胞仪(multiparameter flow cytometry,MFC),最低诊断分型建议参考欧洲白血病免疫分型协作组(EGIL)标准[7],伴髓系表达的标准为肿瘤细胞除表达淋系抗原外同时共同表达任何一种髓系抗原(CD13、CD33、CD14、CD15、MPO)。Ph+ ALL的诊断至少有细胞遗传学或分子生物学一项为阳性,以R显带技术确定染色体核型,核型异常描述体系按人类细胞遗传学国际命名体制(ISCN2005)标准,至少2个细胞具有相同的染色体增加或结构异常、3个细胞具有一致的染色体丢失定义为克隆性染色体异常,复杂核型定义为核型异常涉及3个及以上染色体,并应用RT-PCR检测技术确定BCR-ABL融合基因。 1.3 分组和治疗方法
26例患儿采用CCLG-ALL2008(高危组 HR)方案化疗(称为HR组),27例以TKI联合CCLG-ALL2008(高危组 HR)方案化疗(称为TKI+HR组)。HR组及TKI+HR组未随机,根据患儿病情及监护人意愿在知情同意下进行。
CCLG-ALL2008(高危组 HR)方案参照文献[8]:诊断明确后进行糖皮质激素预治疗,评价治疗反应,之后选择四药联合(长春新碱+柔红霉素+左旋门冬酰胺酶+地塞米松)诱导缓解治疗。TKI+HR组诱导治疗第15天联合应用伊马替尼,第15天、第33天评价骨髓缓解状态并以PCR和MFC法评价微小残留病(MRD),序贯给予CAM×2方案(环磷酰胺+阿糖胞苷+巯嘌呤)早期强化,大剂量甲氨蝶呤等巩固治疗及后期维持治疗,每3个月评价MRD。TKI+HR组根据MRD结果及患儿监护人意愿选择移植或单纯化疗联合伊马替尼,化疗持续期间持续应用伊马替尼(每日260~340 mg/m2)。维持治疗期间每4周中应用伊马替尼2周至停药,后随访观察。伊马替尼停药指征包括:合并严重感染;不良反应严重不能耐受;中性粒细胞减少伴发热。 1.4 安全性评价
美国国立癌症研究所(National Cancer Institute,NCI)化疗毒性分级标准(Common Toxicity Criteria,NCI-CTC 4.0版)评价药物毒性[9]。根据下面的一般准则对每个不良事件的严重程度作特定的临床描述,分为5级。1级为轻度:无症状或轻微;仅为临床或诊断所见;无需治疗。2级为中度:需要较小、局部或非侵入性治疗;与年龄相当的工具性日常生活活动受限。3级为严重或者医学上有重要意义但不会立即危及生命;导致住院或者延长住院时间;致残;个人日常生活活动受限。4级危及生命;需要紧急治疗。5级为与不良事件相关的死亡。 1.5 随访
随访时间至2014年4月30日,失访者截止失访日期(失访5例)。中位随访时间为10个月(1~72个月)。无事件生存(event-free survival,EFS)定义为自诊断到第一次事故(包括复发、CR期间死亡)或末次随访日期。 1.6 统计学分析
以患儿确诊为研究起点,研究终点为死亡、失访或随访截止,数据采用SPSS 17.0统计软件进行处理,符合正态分布的计量资料用均数±标准差(x±s)表示,两组间比较采用成组t检验;偏态分布资料采用中位数和四分位数间距[M(QR)]表示,两组间比较采用秩和检验。计数资料以例数和百分率(%)表示,采用χ2检验比较组间差异。采用Kaplan-Meier方法评估患儿生存率,组间生存率的比较采用log-rank检验。生存分析应用Kaplan-Meier法绘制生存曲线。以P<0.05为有统计学意义的判断标准。 2 结果 2.1 两组临床特征的比较
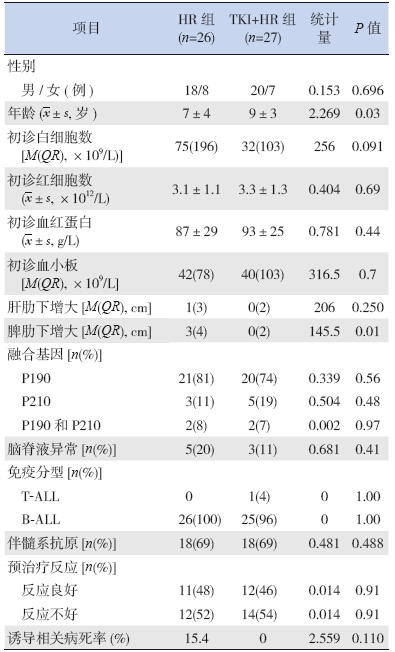
TKI+HR组和HR组的初诊白细胞数、红细胞数、血红蛋白及血小板计数比较差异均无统计学意义。HR组诊断时脾脏增大值大于TKI+HR组,差异有统计学意义。两组患儿免疫表型均以B细胞型为主,B细胞表型中伴髓系表达者均占69%。通过PCR方法检测,患儿以融合转录本BCR-ABL P190阳性为主,HR组及HR+TKI组分别为81%、74%。因此,两组患儿分子生物学的特征基本类似(表 1)。46例患儿进行了细胞遗传学检查,包含有非随机的次要染色体异常者24例,HR组及HR+TKI组分别占57%(12/21)及48%(12/25)。而初诊时51例患儿行脑脊液流式细胞术检测,发现白血病细胞者共8例,两组所占比例分别为20%、11%(P=0.41)。
| 表 1 两组Ph+ ALL患儿临床特征的比较 |
53例患儿总CR率为89%(42/47,去除HR组未评价病例6例),其中进行糖皮质激素预治疗反应评价者共49例,激素治疗反应好者23例,占47%。
HR组患儿共26例,仅以CCLG-ALL2008方案(高危组 HR)序贯治疗,CR率为75%(15/20),未缓解者5例。6例病例无法评价,其原因为:感染相关死亡4例,2例未完成诱导治疗放弃出院。HR组26例患儿中,4例(15%)诱导相关死亡。HR组因早期复发病例多,因此生存分析截止时间较早。HR组3年EFS为(6±5)%(图 1)。
 |
图 1 2HR组和HR+TKl组EFS比较 |
TKI+HR组27例患儿中,诱导期相关病死率为0,CR率为100%(27/27);总体5年EFS为(52±11)%(图 1)。在巩固治疗后11例患儿监护人选择移植治疗,移植组3年EFS为(64±15)%。移植患儿中,1例为同胞供者造血干细胞移植,3例为无关供者造血干细胞移植(移植相关并发症死亡1例),7例为半相合造血干细胞移植(移植相关并发症死亡2例,移植后复发死亡1例)。无关供者和半相合造血干细胞移植组3年EFS分别为(67±27)%、(57±19)%(P=0.90)(图 2)。化疗联合靶向治疗组患儿共16例(其中6例患者由于经济原因中途放弃伊马替尼及化学药物治疗),2例患者复发(1例为维持治疗期间单纯中枢神经系统复发,1例为巩固强化治疗期间出现ABL激酶ST315I突变后复发),此组患儿5年EFS为(53±19)%。总体而言,在TKI+HR组中,移植和化疗患儿之间EFS差异无统计学意义(P=0.69)(图 3)。
 |
图 2 HR+TKl组不同供者来源EFS比较 |
 |
图 3 HR+TKl组移植和化疗患儿EFS比较 |
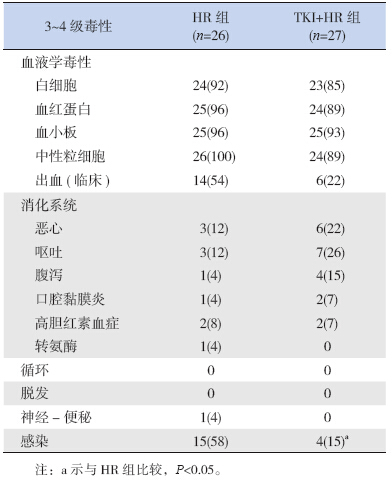
对HR组及TKI+HR组初诊诱导缓解过程中化疗药物相关毒性进行评价,发现伊马替尼的应用未增加化疗相关毒性,不良反应主要表现为恶心、呕吐、腹泻、脱发、胆红素升高等,但大多为1~2级,对症治疗可好转;两组患儿的血液学毒性均为3~4级,两组比较发生率基本相当;TKI+HR组感染发生率明显低于HR组(P=0.022)(表 2)。
| 表 2 伊马替尼联合CCLG-ALL2008方案在诱导缓解期3~4级毒性反应评价[n(%)] |
Ph染色体是22号染色体部分(22q11)易位到9号染色体上(9q34.11)所形成,C-abl原癌基因位于第9号染色体的断裂点上,其翻译产物P145蛋白具有内在络氨酸活性,而定位于人类第22号染色体的bcr,常见有3个断裂点区域,即M-bcr、m-bcr及u-bcr,从而形成不同的bcr/abl融合转录本及其蛋白产物(即P210、P190、P230蛋白)。Ph+ ALL见于7%~16%的青少年ALL和2%~3%儿童ALL[1, 2],其蛋白产物大多为P190,具有发病年龄较大,起病时白细胞数高,且随年龄增长发病呈升高趋势等特点,以上多个因素导致此亚型患儿预后极差[10, 11]。TKI的出现,使儿童Ph+ ALL的疗效及其治疗策略发生了很大的改变。本研究比较我中心应用TKI与否的儿童Ph+ ALL,无论是CR率、诱导相关病死率,还是长期生存率,均有显著差异。
靶向药物TKI应用之前,多数研究建议儿童Ph+ ALL应在CR1期尽早行异基因造血干细胞移植。强诱导化疗后Ph+ ALL CR率在70%~90%间,但大部分患者在治疗12个月内复发或死亡[12]。Arico等[13]总结1986~1996年期间326例儿童Ph+ ALL患者,中位年龄8.1岁,CR率为82%,化疗组和移植组的7年EFS分别为(25±4)%、(65±8)%;7年OS分别为(42±4)%、(72±8)%;之后Arico等[14]再次对后10年间(1995~2005年)的儿童Ph+ ALL患者(共610例)进行研究,其CR率为89%,化疗组和移植组的7年EFS分别为(34.2±4)%、(44±2.9)%;7年OS分别为(44±3)%、(54±2.8)%;对比而言,虽然有治疗手段的进步,但是前后10年比较而言,临床疗效提高并不明显,同时研究也提示相关供者移植和无关供者移植之间的疗效无显著性差异。因此,TKI时代来临之前,移植是儿童Ph+ ALL治疗的一线选择。
随着靶向药物TKI的应用,儿童Ph+ ALL治疗有了长足的进步,国际多中心研究COG-AALL0031方案采取强化疗联合伊马替尼治疗儿童Ph+ALL,CR率达95%,而诱导相关病死率为0;同时在AALL0031方案中对伊马替尼的应用方式和疗程进行了多层次的长期观察,研究表明在化疗同时持续应用伊马替尼可获得更好的疗效,强化疗联合伊马替尼组5年DFS可达(70±12)%,同胞供者和无关供者造血干细胞移植组5年DFS分别为(65±11)%、(59±15)%[3, 4, 5];同期,欧洲多个儿童Ph+ ALL协作组(EsPhALL)以及西班牙(SEHOP/SHOP)儿童白血病协作组关于伊马替尼的随机对照研究,均证明伊马替尼治疗使Ph+ ALL儿童获益,同时与化疗联合应用,具有良好的安全性[15, 16]。
本研究病例具有年长儿居多、初诊白细胞数高等特点,主要为B细胞表型,伴有髓系表达者占69%,与国外报道类似[15, 16]。比较HR及TKI+HR两组疗效发现,HR组的CR率相对较低,仅为75%,且诱导相关病死率很高;而TKI+HR组,其CR率达到100%,无诱导相关死亡病例且诱导期死亡的主要原因——感染的发生率明显降低。由于诱导失败、诱导相关死亡、早期复发等多方面原因,HR组3年EFS仅为(6±5)%。而TKI+HR组患儿,其整体疗效获得提高,总体5年EFS达到(52±11)%。TKI+HR组的移植患儿中,由于我国的国情特点,选择半相合造血干细胞移植的病例数较多,但无关供者和半相合造血干细胞移植组之间的生存率无显著性差异。总体而言,尽管病例数有限,治疗方法多样,但TKI+HR组5年总体生存率较HR组明显提高。
有报道称,总结AALL0031方案强化疗组在儿童Ph+ALL患儿中之所以产生如此优越疗效的原因,除了儿童ALL化疗方案的优点,如化疗总疗程持续时间长、大剂量甲氨蝶呤及门冬酰胺酶的应用、中枢神经系统白血病的预防、维持化疗时间长等多方面的因素有关外,伊马替尼的治疗时间长应当是其最大的特点[10],因此,对于儿童Ph+ ALL治疗方案的优化仍是值得探索的目标。
COG同样评价了伊马替尼在儿童患者中的安全性,COG-2004方案选择不同剂量伊马替尼单药治疗儿童Ph+ ALL,不良反应主要包括恶心(4%)、呕吐(3.5%)、乏力、嗜睡、心悸(3.5%)、肝脏转氨酶升高(6.7%)、腹泻(2.7%)、关节痛/肌痛(1.5%)、水肿/体重增加(不足1%),多表现为1~2级,均较轻微[17]。而欧洲EsPhALL以及西班牙SEHOP/SHOP协作组总结伊马替尼联合化疗在儿童Ph+ ALL的诱导缓解和巩固治疗中的毒性亦未见明显增加[15, 16]。同样,本研究总结了两组患儿在诱导缓解治疗中的化疗相关毒性,TKI+HR组不良反应主要表现为恶心、呕吐、腹泻、脱发、胆红素升高等,但大多为1~2级;而对于明显的血液学毒性TKI+HR组并未增加,相反,其血液学毒性相关感染的发生率却明显降低,可能与其诱导缓解率升高,完全的血液学反应有助于感染的控制有关系。
儿童Ph+ ALL患者治疗方案的选择目前存在很大的争议,无论移植还是化疗,可能需要MRD的监测等多方面的因素共同决定[5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18]。本研究显示应用CCLG-ALL2008方案联合TKI治疗儿童Ph+ALL在疗效上发生了很大的变化,但是在方案的实施过程中,由于多方面原因的限制,也存在了许多问题,例如:很多患儿不能持续应用TKI,部分患儿中途放弃治疗,或者不能进行有效的MRD的监测而失去选择更有效和适合的治疗方案的机会等。同时,关于TKI的耐药问题以及二代TKI的临床应用,均需要进一步完善。总体说来,TKI的出现使儿童Ph+ ALL的治疗疗效获得明显改善,同时具有良好的安全性。
| [1] | Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia[J]. N Engl J, 2006, 354(2): 166-178. |
| [2] | Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia[J]. N Engl J Med, 2000, 342(14): 998-1006. |
| [3] | Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study[J]. J ClinOncol, 2009, 27(31): 5175-5181. |
| [4] | Schultz KR, Bowman WP, Aledo A, et al. Continuous dosing imatinib with intensive chemotherapy gives equivalent outcomes to allogeneic BMT for Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL) with longer term follow up: Updated Results of Children's Oncology Group (COG)AALL0031[J]. Pediatr Blood Cancer, 2010, 54: 788. |
| [5] | Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group Study AALL0031[J]. Leukemia, 2014, 28(7): 1467-1471. |
| [6] | 中华医学会儿科学分会血液学组. 2014 儿童急性淋巴细胞白血病诊疗建议(第四次修订)[J]. 中华儿科杂志, 2014, 52(9): 641-644. |
| [7] | Bene MC, Castoldi G, Knapp W, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL)[J]. Leukemia, 1995, 9(10): 1783-1786. |
| [8] | 陈晓娟, 邹尧, 杨文钰, 等. CCLG-ALL2008方案治疗儿童急性淋巴细胞白血病复发患儿的特征分析[J]. 中国当代儿科杂志, 2015, 17(4): 321-326. |
| [9] | Common Terminology Criteria for Adverse Events (CTCAE) v4.0 (CTCAE) [S][v4.03: June 14, 2010]. U.S. Department of Health and Human Services. National Institutes of Health National Cancer Institute, 2008. |
| [10] | Schultz KR, Prestidge T, Camitta BT. Philadelphia chromosome-positive acute lymphoblastic leukemia in children: new and emerging treatment options[J]. Expert Rev Hematol, 2010, 3(6): 731-742. |
| [11] | Arico M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia[J]. N Engl J Med, 2000, 342(14): 998-1006. |
| [12] | Lee HJ, Tompson JE, Wang ES, et al. Philadelphia chromosome-positive acute lymphoblastic leukemia: current treatment and future perspective[J]. Cancer, 2011, 117(8): 1583-1594. |
| [13] | Arico M, Valsecchi MG, Conter V, et al. Improved outcome in high-risk childhood acute lymphoblastic leukemia defined by prednisonepoor response treated with double Berlin-Frankfurt-Muenster protocol II[J]. Blood, 2002, 100(2): 420-426. |
| [14] | Aricò M, Schrappe M, Hunger SP, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005[J]. J Clin Oncol, 2010, 28(31): 4755-4761. |
| [15] | Rives S, Estella J, Gómez P, et al. Intermediate dose of imatinib in combination with chemotherapy followed by allogeneic stem cell transplantation improves early outcome in paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (ALL): results of the Spanish Cooperative Group SHOP studies ALL-94, ALL-99 and ALL-2005[J]. Br J Haematol, 2011, 154(5): 600-611. |
| [16] | Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study[J]. Lancet Oncol, 2012, 13(9): 936-945. |
| [17] | Champagne MA, Capdeville R, Krailo M, et al. Imatinibmesylate (STI571) for treatment of children with Philadelphia chromosome-positive leukemia: results from a Children's Oncology Group phase 1 study[J]. Blood, 2004, 104(9): 2655-2660. |
| [18] | Jeha S, Coustan-Smith E, Pei D, et al. Impact of tyrosine kinase inhibitors on minimal residual disease and outcome in childhood Philadelphia chromosome-positive acute lymphoblastic leukemia[J]. Cancer, 2014, 120(10): 1514-1519. |