 2015, Vol. 17
2015, Vol. 17



慢性缺氧引起的肺血管收缩和血管重塑是肺动脉高压的主要病理基础,肺动脉高压的形成和体内氧化还原失衡有关[1, 2]。缺氧使得机体处于氧化应激状态,活性氧簇(reactive oxygen species,ROS)大量产生。研究表明ROS是低氧性肺血管收缩及重塑的重要上游信号分子。各种类型的细胞都可生成ROS,包括内皮细胞、平滑肌细胞及外膜细胞等[3, 4]。烟酰胺腺嘌呤二核苷酸磷酸(nicotinamideadenine dinucleotide phosphate,NADPH)氧化酶,简称NADPH氧化酶(NADPH oxidase,NOX)是血管内ROS生成的最主要酶体,也是体内重要的氧感受器[5, 6]。近年来非吞噬细胞NOX的研究受到重视,在不同种类细胞中发现一系列NOX催化亚单位,目前认为它参与了多种病理生理过程。Li等[7]发现NOX4在低氧性肺动脉高压形成过程中起到了重要的作用。多项实验表明NOX2和NOX4参与了低氧性肺血管收缩和重塑[8, 9]。
内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)也可参与ROS的生成,在氧化应激状态下,eNOS功能出现异常,合成的终产物为ROS非NO,而且ROS可降低NO的生物学活性,从而加剧内皮功能的异常[10]。Dikalova等[11]发现eNOS和NOX之间有着重要的关联。NOX活化导致ROS的过度生成,ROS可抑制eNOS合成NO而是生成更多的过氧化离子,从而推测eNOS和NOX之间存在平衡制约关系。目前关于eNOS在低氧性肺动脉高压过程中对NOX的调节作用,尤其是体内NO缺乏情况下NOX表达及ROS的变化尚无研究。
4-羟基-2,2,6,6-四甲基哌啶(TEMPOL)是一种膜通透性的超氧化物歧化酶,可以直接清除机体内的活性氧并能选择性降低肺动脉高压[12]。本研究利用它的这一特性,研究了野生型及eNOS基因敲除小鼠在慢性缺氧条件下及TEMPOL干预后NOX2和NOX4 mRNA表达及肺组织ROS浓度的变化。
1 材料与方法 1.1 实验动物及分组清洁级健康成年雄性C57BL/6小鼠购自浙江大学医学院动物中心,eNOS基因敲除健康成年雄性C57BL/6小鼠购自美国Jackson实验室(编号002684)。基因敲除小鼠实验前进行了基因验证。两种小鼠各挑选30只,体重18~22 g,随机分成5组,每组每种小鼠各6只。(1)常氧组:将成年C57BL/6小鼠置于自行设计的密封有机玻璃箱中,维持FiO2在21%左右,21 d后处死。(2)低氧组:将成年C57BL/6小鼠置于FiO2为10%左右的密封有机玻璃箱中饲养7 d (低氧7 d组)或21 d (低氧21 d组)后处死。(3)低氧+治疗组:成年C57BL/6小鼠在FiO2为10%左右的密封有机玻璃箱中饲养,在其饮用水中加入TEMPOL (10 mmol/L)干预7 d (治疗7 d组)或21 d (治疗21 d组)后处死。饲养过程保持箱内温度25~27℃,湿度50%~70%,每3 d开箱半小时更换水、食物及垫料。
1.2 肺组织分离及右心室肥厚指标测定各组小鼠放血处死后,分离肺组织,用生理盐水进行肺动脉灌洗,直到流出液转清,其左肺置于液氮中,-80℃保存,用于实时荧光定量多重聚合酶链反应(RT-PCR)检测;右肺用4%多聚甲醛固定后,石蜡包埋,用于免疫组化及苏木精-伊红(HE)染色。剪去心房和游离的大血管,沿着房室间隔剪下右心室壁,分别称量右心室(RV)、左心室(LV)和室间隔(S)的重量,结果以RV/(LV+S)表示。
1.3 免疫组化法检测肺血管重塑各组肺组织标本用SMA抗体免疫标记后,应用Image Pro Plus对肺组织切片中内弹力层清晰、平滑肌层完整、形状较规则且直径在80~120 μm之间的肺细小动脉膜厚度和管腔直径进行测定,用血管中膜的厚度与管腔直径的比值MT%评价血管重塑情况。每只小鼠留取3张切片,每张切片随机选取3~5个视野,每个视野选择3~5个血管。
1.4 肺组织ROS浓度的测定取肺组织约0.1 g,以1:20(重量体积比)的比例加入冰冷的0.1 mol/L磷酸钾缓冲液(pH 7.4),用玻璃匀浆器在0~4℃下制成匀浆液,将匀浆液在4℃下以1 000 g离心10 min后取上清液;加入1 mmol/L DCFH-DA,混匀,置于酶标仪中37℃温浴30 min,以485 nm波长为激发光,于53 nm波长处测荧光强度(OD)值。
1.5 RT-PCR检测NOX2、NOX4及eNOS的mRNA表达采用AXYPrep总RNA小量制备试剂盒抽提肺组织总RNA,应用宝生物公司反转录试剂盒逆转录为cDNA,引物序列设计见表 1。用荧光定量PCR检测NOX2、4和eNOS的mRNA表达量。PCR反应体系:CDNA模板2 µL,上下游引物各0.5 µL,SYBR Premix Ex TaqTM II 12.5 µL,ddH2O补足体积至25 µL。PCR反应条件:94℃预变性10 s;94℃变性15 s,60℃退火50 s,重复40个循环。设定在每个循环的退火期结束后程序自动记录该循环的荧光值,以表示在该循环结束时PCR产物的量。某样本目的基因mRNA的表达量以⊿ Ct表示,⊿ Ct=Ct (目的基因)-Ct (内参基因),⊿ Ct越小,该样本目的基因mRNA的表达量越大。
| 表 1 引物序列设计 |

采用SPSS 16.0统计软件对数据进行统计学分析,计量资料采用均数±标准差(x±s)表示,多组间比较采用单因素方差分析,组间两两比较采用SNK-q检验,P<0.05为差异有统计学意义。
2 结果 2.1 各组肺血管重塑及右心室肥厚指标的变化各组野生型小鼠MT%比较差异有统计学意义(F=15.16,P<0.01);其中低氧21 d组MT%(24.9±4.0)明显高于常氧组(14.3±1.3)和治疗21 d组(23.2±4.6)(P<0.05);常氧组和治疗21 d组比较差异无统计学意义(P>0.05)。各组基因敲除小鼠MT%比较差异有统计学意义(F=9.41,P<0.01);其中低氧21 d组MT%(23.3±4.4)显著高于常氧组(15.4±2.0)和治疗21 d组(22.6±3.6)(P<0.05);常氧组和治疗21 d组比较差异无统计学意义(P>0.05)。见图 1。
 |
图 1 野生型和基因敲除小鼠缺氧及治疗后肺血管重塑情况(α-SMA 免疫组化,×400) 棕色染色表示 α-SMA染色阳性,可反映血管中膜厚度;蓝色染色代表细胞核。 |
各组野生型小鼠RV/(LV+S)比值比较差异有统计学意义(F=29.14,P<0.01);其中低氧21 d组RV/(LV+S)比值(0.353±0.036)明显高于常氧组(0.246±0.026)和治疗21 d组(0.267±0.038)(P<0.01);常氧组和治疗21 d组比较差异无统计学意义(P>0.05)。各组基因敲除小鼠RV/(LV+S)比值比较差异有统计学意义(F=16.42,P<0.01),其中低氧21 d组RV/(LV+S)比值(0.379±0.039)高于常氧组(0.233±0.022)和治疗21 d组(0.302± 0.033)(P<0.01);常氧组和治疗21 d组比较差异无统计学意义(P>0.05)。见图 2。
 |
图 2 野生型和基因敲除小鼠缺氧及治疗后右心室肥厚指标的变化 RV:右心室;LV:左心室;S:室间隔;a 示与同种小鼠常氧组比较, P<0.01,b 示与同种小鼠低氧 21 d 组比较,P<0.01。 |
野生型小鼠各组肺组织ROS的浓度比较差异有统计学意义(F=42.4,P<0.01);其中低氧21 d组(264±97 u)和治疗21 d组ROS浓度(201±106 u)与常氧组(1 000±290 u)比较均明显下降(P<0.01),治疗21 d组与低氧21 d组ROS浓度比较差异无统计学意义(P>0.05)。基因敲除小鼠常氧组ROS的浓度(688±262 u)与同组野生型小鼠比较显著下降(P<0.05)。基因敲除小鼠各组肺组织ROS浓度比较差异无统计学意义(F=3.05,P=0.077)。见图 3。
 |
图 3 野生型和基因敲除小鼠缺氧及治疗后肺组织ROS 浓度的变化 a 示与同种小鼠常氧组比较, P<0.01,b 示与同组野生型小鼠比较, P<0.05。 |
野生型小鼠各组NOX2 mRNA的表达差异有统计学意义(P<0.01),其中低氧组NOX2 mRNA的表达水平明显高于常氧组(P<0.01);治疗7 d组NOX2 mRNA的表达水平低于低氧7 d组(P<0.01)。野生型小鼠各组NOX4 mRNA的表达差异有统计学意义(P<0.01),其中低氧组NOX4 mRNA的表达水平高于常氧组(P<0.01);治疗7 d组和治疗21 d组NOX4 mRNA的表达水平分别低于低氧7 d组和低氧21 d组(P<0.05)。野生型小鼠各组eNOS mRNA的表达差异有统计学意义(P<0.01),其中低氧组eNOS mRNA的表达水平高于常氧组(P<0.01);治疗7 d组eNOS mRNA的表达水平低于低氧7 d组(P<0.01)。见表 2。
| 表 2 各小组肺组织 NOX2、 NOX4 和 eNOS mRNA 的表达(n=6,x±s) |
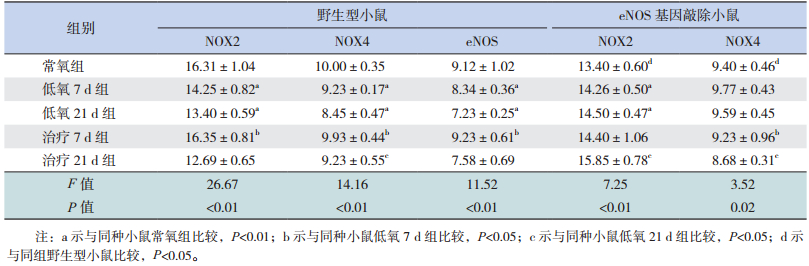
基因敲除小鼠各组NOX2 mRNA的表达差异有统计学意义(P<0.01),其中常氧组NOX2 mRNA表达水平显著高于低氧组和同组野生型小鼠(P<0.01);治疗21 d组NOX2 mRNA的表达水平低于低氧21 d组(P<0.05)。基因敲除小鼠各组NOX4 mRNA的表达水平差异有统计学意义(P<0.05),其中常氧组NOX4 mRNA表达水平显著高于同组野生型小鼠(P<0.05);治疗7 d组和治疗21 d组NOX4 mRNA的表达水平分别较低氧7 d组和低氧21 d组显著上升(P<0.05)。见表 2。
3 讨论本实验中结果发现野生型和eNOS基因敲除小鼠慢性缺氧可导致肺血管重塑及右心室肥厚,TEMPOL干预可部分逆转上述改变。NOX是肺血管ROS的主要来源。Liu等[13]发现NOX2基因敲除的小鼠缺氧后肺血管重塑情况比野生型小鼠轻。Mittal等[14]发现NOX蛋白家族中NOX4在小鼠肺组织中的表达最高。小鼠慢性缺氧后肺组织NOX4 mRNA和蛋白的表达随着缺氧时间的延长逐步升高,与慢性缺氧肺血管重塑的发展过程一致[15]。NOX4在人肺动脉平滑肌细胞中广泛表达,肺动脉高压患者肺组织NOX4基因及蛋白表达明显高于正常人群[16]。这和本实验结果一致。本实验结果中发现缺氧可诱导野生型小鼠肺组织NOX2和NOX4 mRNA的过度表达,且随缺氧时间的延长逐步增加,提示缺氧后机体一直处于氧化应激状态。从这一途径而言,体内ROS的含量应该明显增加。但是由于整个机体内ROS浓度与局部组织中的ROS含量并不一致,所以缺氧后肺组织ROS的含量是上升还是下降一直有争议[17]。本实验中结果发现野生型小鼠缺氧后肺组织ROS的含量下降,这与NOX的表达趋势相反。对于这个矛盾的结果,我们认为首先NOX基因表达上升和其NOX活性上升并不一致;其次,NOX仅仅是ROS的主要来源之一;此外,ROS的浓度不仅和ROS的合成有关,还和它的清除密切相关。目前研究认为NOX和eNOS之间有着密切的关系,NOX来源的ROS会导致NOS "uncoupling" ,使得其活性改变。Sanchez等[18]报道下调NOX的表达,会增加eNOS的活性,降低血管压力。本研究首次研究在NO缺乏的情况下且缺氧条件下NOX和ROS的变化。在基因敲除的小鼠中,肺组织NOX2 mRNA的表达明显高于野生型,NOX4 mRNA的表达略高于野生型小鼠;肺组织ROS的浓度比野生型小鼠低。我们推测在体内eNOS来源的NO缺乏情况下,体内NO及NOX的ROS之间的平衡被破坏,推测eNOS对NOX的表达有一定的抑制作用。基因敲除小鼠缺氧后NOX2 mRNA的表达下降而NOX4 mRNA及肺内ROS的浓度无明显变化,因此eNOS可能是NOX家族中NOX2基因表达的重要调控因素,或者两者之间的表达存在某种负反馈机制。所以,对于基因敲除小鼠,NOX2在肺动脉形成过程中起到了更重要的作用,低氧性肺血管重塑也可能是通过其他途径实现的。
研究表明ROS是低氧性肺血管收缩及重塑的上游信号分子,在低氧早期,它是肺血管重塑的级联反应的起始分子。Victor等[19]发现缺氧早期基因敲除小鼠肺小血管肌化程度明显少于野生型小鼠。我们认为这一改变和NOX2、4的表达下降有关。文献表明TEMPOL可降低细胞内ROS的浓度,选择性降低肺动脉压力。本研究结果发现TEMPOL除了清除体内过度生成的ROS,还可以降低NOX2和NOX4的基因表达,这个作用在缺氧早期更明显。实验结果发现eNOS的变化和NOX的变化相一致,因此推测两者的表达是相互制约的。对于基因敲除小鼠,TEMPOL干预可降低RV/(LV+S)的比值。这和Hodyc等[20]研究小组的结果相吻合。但确切的机制尚不清楚。
本研究认为低氧条件下小鼠肺组织eNOS和NOX2、4的基因表达之间有着重要的关联,eNOS对NOX的表达有重要的调节作用。由于本实验仅在基因水平做了探讨,关于以上分子的蛋白质的表达及酶活性尚待进一步研究。致谢:感谢浙江大学附属儿童医院内科实验室沈征、舒小丽对本实验的帮助。
| [1] | Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension[J]. Circ Res, 2014, 115(1):148-164. |
| [2] | 齐建光, 邢长青, 丁亚光, 等. 肾上腺髓质素缓解低氧性肺 动脉高压大鼠肺动脉中胶原堆积的研究[J]. 中国当代儿科杂 志, 2012, 14(1):54-58. |
| [3] | Aggarwal S, Gross CM, Sharma S, et al. Reactive oxygen species in pulmonary vascular remodeling[J]. Compr Physiol, 2013, 3(3):1011-1034. |
| [4] | MacKay CE, Knock GA. Control of vascular smooth muscle function by Src-family kinases and reactive oxygen species in health and disease[J]. J Physiol, 2014. [Epub ahead of print]. |
| [5] | Schulz R, Murzabekova G, Egemnazarov B, et al. Arterial hypertension in a murine model of sleep apnea:role of NADPH oxidase 2[J]. J Hypertens, 2014, 32(2):300-305. |
| [6] | Chen F, Barman S, Yu Y, et al. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species[J]. Free Radic Biol Med, 2014, 73:201-213. |
| [7] | Li S, Tabar SS, Malec V, et al. NOX4 regulates ROS levels under normoxic and hypoxic conditions, triggers proliferation, and inhibits apoptosis in pulmonary artery adventitial fibroblasts[J]. Antioxid Redox Signal, 2008, 10(10):1687-1698. |
| [8] | Frazziano G, Al Ghouleh I, Baust J, et al. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension:indications for a seminal role for mitochondrial Nox4[J]. Am J Physiol Heart Circ Physiol, 2014, 306(2):H197-H205. |
| [9] | Norton CE, Broughton BR, Jernigan NL, et al. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2[J]. Antioxid Redox Signal, 2013, 18(14):1777-1788. |
| [10] | Harrison IP, Selemidis S. Understanding the biology of reactive oxygen species and their link to cancer:NADPH oxidases as novel pharmacological targets[J]. Clin Exp Pharmacol Physiol, 2014, 41(8):533-542. |
| [11] | Dikalova AE, Góngora MC, Harrison DG, et al. Upregulation of Nox1 in vascular smooth muscle leads to impaired endotheliumdependent relaxation via eNOS uncoupling[J]. Am J Physiol Heart Circ Physiol, 2010, 299(3):H673-H679. |
| [12] | Rashid M, Kotwani A, Fahim M. Long-acting phosphodiesterase 5 inhibitor, tadalafil, and superoxide dismutase mimetic, tempol, protect against acute hypoxia-induced pulmonary hypertension in rats[J]. Hum Exp Toxicol, 2012, 31(6):626-636. |
| [13] | Liu JQ, Zelko IN, Erbynn EM, et al. Hypoxic pulmonary hypertension:role of superoxide and NADPH oxidase (gp91phox)[J]. Am J Physiol Lung Cell Mol Physiol, 2006, 290(1):L2-L10. |
| [14] | Mittal M, Gu XQ, Pak O, et al. Hypoxia induces Kv channel current inhibition by increased NADPH oxidase-derived reactive oxygen species[J]. Free Radic Biol Med, 2012, 52(6):1033-1042. |
| [15] | Barman SA, Chen F, Su Y, et al. NADPH oxidase 4 is expressed in pulmonary artery adventitia and contributes to hypertensive vascular remodeling[J]. Arterioscler Thromb Vasc Biol, 2014, 34(8):1704-1715. |
| [16] | Lu X, Murphy TC, Nanes MS, et al. PPAR{gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B[J]. Am J Physiol Lung Cell Mol Physiol, 2010, 299(4):L559-L566. |
| [17] | Fuchs B, Sommer N, Dietrich A, et al. Redox signaling and reactive oxygen species in hypoxic pulmonary vasoconstriction[J]. Respir Physiol Neurobiol, 2010, 174(3):282-291. |
| [18] | Sanchez M, Galisteo M, Vera R, et al. Quercetin downregulates NADPH oxidase, increases eNOS activity and prevents endothelial dysfunction in spontaneously hypertensive rats[J]. J Hypertens, 2006, 24(1):75-84. |
| [19] | Victor VM, Nuñez C, D'Ocón P, et al. Regulation of oxygen distribution in tissues by endothelial nitric oxide[J]. Circ Res, 2009, 104(10):1178-1183. |
| [20] | Hodyc D, Snorek M, Brtnicky T, et al. Superoxide dismutase mimetic tempol inhibits hypoxic pulmonary vasoconstriction in rats independently of nitric oxide production[J]. Exp Physiol, 2007, 92(5):945-951. |