 2015, Vol. 17
2015, Vol. 17



维生素D受体(vitamin D receptor,VDR)是介导维生素D发挥生物效应的核内大分子蛋白,属于类固醇激素/甲状腺激素受体超家族成员。研究发现VDR除了调节钙磷代谢的经典作用以外,还有其他重要功能:VDR能通过影响肿瘤细胞的增殖和凋亡影响恶性肿瘤疾病的预后[1];VDR与维生素D结合后能抑制肾素-血管紧张素系统,从而影响心血管疾病的进程[2];此外,越来越多的研究发现VDR参与免疫炎性调控[3]。
VDR除了在常见的皮肤、骨骼、胃肠道等组织表达以外,也表达于单核细胞、巨噬细胞、树突状细胞和活化的T淋巴细胞等免疫细胞。研究发现,VDR参与炎性肠病、哮喘、糖尿病、过敏性紫癜、慢性肾病和风湿性疾病等疾病的病程[4, 5],越来越多的研究提示VDR及其配体维生素D在疾病的炎症和免疫过程中发挥了非常重要的作用[6]。已知病毒性心肌炎(viral myocarditis,VMC)是由病原体引起的心肌炎性免疫性疾病,VDR在VMC病程中的作用,国内外少有相关报道。本实验通过建立小鼠VMC模型,动态观察和探讨VDR在VMC小鼠心肌中的表达变化及其作用,并探究VDR表达水平与心肌病理变化程度之间的相关性,旨在为VMC的临床诊疗工作带来新的理论依据。
1 材料与方法 1.1 病毒准备安徽医科大学微生物实验室提供的柯萨奇B3病毒(coxsackievirus B3,CVB3)在Hep-2细胞上传代、扩增,添加含有2%胎牛血清的细胞维持液培养,镜下观察,48 h后当细胞病变达到75%时收集病毒液冷冻。将冷冻的病毒液反复冻融3次以充分释放病毒颗粒,4℃以2 000 rpm离心10 min,取其上清液分装后-70℃冻存备用。在96孔板接种生长良好的Hep-2细胞,添加含有10%胎牛血清的细胞培养液,置于37℃、5% CO2条件下培养,待细胞将孔底铺满时,取一小支病毒液10倍稀释,在96孔板上滴定病毒的半数组织感染剂量(50% Tissue culture infective dose,TCID50):显微镜下观察细胞的病变情况,当细胞折光性降低,细胞皱缩、脱落浮起时视为病变,逐日监测细胞病变情况一周,观察病变细胞> 50%的板孔,用Read-Muench双氏法计算出CVB3的TCID50为10-7。
1.2 材料兔来源多克隆抗体购自英国abcam公司;SP法二抗试剂盒购自北京中山金桥生物技术有限公司;磷酸盐缓冲液(PBS)购自武汉博士得生物有限公司。
1.3 实验动物与取材从安徽医科大学实验动物中心购得120只4周龄雄性SPF级BALB/c小鼠,体重14~18 g,按照随机数字表法设立实验组小鼠80只,对照组40只。碘伏消毒小鼠腹部,每只实验组小鼠腹腔注射0.1 mL含有CVB3 1×102 TCID50的上清液,对照组小鼠则注射0.1 mL的细胞培养液DMEM。将小鼠饲养于清洁级环境中,密切观察小鼠的饮食量、毛色与掉毛情况、精神状况,并记录小鼠死亡情况。分别于注射后第3、7、14和28天随机各取实验组小鼠15只,对照组小鼠10只,称其体重,观察比较实验组与对照组体重变化情况。将小鼠麻醉、眼球取血处死,取得小鼠心脏组织,剪取适量大小于10%中性甲醛中固定,以备用于免疫组织化学以及组织病理学检测。
1.4 免疫组织化学技术检测VDR表达将在中性甲醛中固定的心肌组织脱水、石蜡包埋制成蜡块;4 μm厚度连续切片,用经多聚赖氨酸防脱处理的载玻片捞片,于65℃温箱烤片3 h;二甲苯脱蜡,无水酒精、95%、85%、75%酒精梯度水化;滴加3%过氧化氢溶液覆盖组织10 min,以阻断组织内源性过氧化氢酶,PBS冲洗5 min重复3次;柠檬酸盐浸泡微波抗原修复10 min后,停1 min加热1 min反复5次,自然冷却至室温;PBS冲洗后滴加山羊血清封闭15 min,甩掉血清,滴加一抗,一抗为兔来源多克隆抗体,经预实验确定一抗最佳稀释浓度为1:3 500,同时用PBS替代一抗覆盖组织作为阴性切片,4℃过夜。37℃复温30 min,冲洗后滴加二抗37℃孵育20 min;冲洗后滴加试剂盒中的辣根酶标记链霉卵白素,37℃孵育20 min,PBS冲洗;避光配置DAB,滴加DAB显色,镜下观察并控制显色时间,显色时间为3~5 min,双蒸水冲洗终止显色;苏木精复染15 s,冲洗、温水浸泡返蓝,梯度酒精脱水,中性树脂封片。
每个标本随机选取3张切片,每张切片随机选取5个无重叠高倍镜(×400)视野,由与本实验无关的两位病理学研究人员计数视野中阳性细胞数、阳性细胞率,并观察染色强度。按照细胞阳性率的多少,评分为0~4分:<5%为0分,5%~20%为1分,21%~50%为2分,51%~70%为3分,71%~100%为4分;按照染色深浅程度,评分为1~3分:淡黄色为1分,棕黄色为2分,深褐色为3分;将两种评分相加并算出平均分[7]。
1.5 组织病理学检测烤片、脱蜡水化如免疫组化步骤,苏木精染色8 min,冲洗,分化返蓝,伊红染色1 min,脱水,中性树脂封片。显微镜下观察心肌组织病理变化,每个标本随机选取3张切片,每张切片随机选择5个高倍镜视野(×400),按照视野中炎性细胞浸润和炎症坏死区域面积与整个视野面积之比评分为0~4分[8]:无坏死和炎症细胞浸润评为0分,坏死、炎性细胞浸润面积占总面积<25%评为1分,坏死、浸润面积达到25%~50%时评为2分,51%~75%时评为3分,>75%时评为4分。
1.6 统计学分析使用SPSS 16.0统计软件对数据进行统计学分析,正态分布计量资料采用均数±标准差(x±s)表示,两独立样本均数的比较采用t检验;多组计量资料的比较采用单因素方差分析,组间两两比较采用SNK-q检验。P<0.05为差异有统计学意义。
2 结果 2.1 实验动物一般情况实验组小鼠3 d后毛色暗淡无光泽,开始出现掉毛,体重不增甚至下降,活动量明显下降,饮食量减少。观察到实验组小鼠死亡19只,死亡率为24%,第7天左右为小鼠死亡高峰期,14 d以后没有小鼠死亡。对照组小鼠毛色光亮,无明显脱毛现象,饮食、活动情况良好,没有小鼠死亡。
2.2 心肌标本组织病理学变化实验组小鼠在第7天和第14天心脏大体解剖发现,心脏外膜出现明显的、大小不等的坏死病灶,肉眼观察呈地图状白斑,对照组小鼠心脏未见白斑;第28天时,实验组小鼠心脏比对照组明显增大。
HE染色对实验组小鼠心肌组织进行组织病理学观察,结果如图 1所示,在第3天,小鼠心肌细胞排列不规则,可见少量炎症细胞浸润;第7天时,小鼠心肌组织大量破坏,心肌细胞增粗肿胀、排列严重紊乱,出现大片坏死灶,坏死区域细胞膜、胞核破裂瓦解,周围可见大量炎症细胞浸润;第14天,心肌局部炎症、坏死病灶可见吸收,但心肌仍然可见大量坏死区域和炎症细胞浸润;第28天,心肌大部分炎症坏死病灶已吸收,炎症细胞明显减少,大量纤维细胞增生,心肌细胞呈代偿性肥大状态。
 |
图 1 小鼠心肌 HE 染色变化(×400) A:正常对照组心肌排列整齐,未见炎症、坏死病灶;B:实验组第 3 天,心肌排列不规则,可见少量炎症细胞浸润;C:实验组第 7 天,心肌排列紊乱,可见大量炎症细胞浸润和大片坏死病灶(箭头所示);D:实验组第 14 天,心肌组织仍可见较多炎性细胞浸润和坏死病灶;E:实验组第 28 天,心肌炎症坏死病灶大部分已被吸收。 |
经统计分析,在第3、7、14和28天,实验组小鼠心肌组织病理积分分别为1.48±0.27、3.49±0.26、2.51±0.23和1.05±0.26,均高于正常对照组(0)(分别t=21.19、51.96、43.13和15.93,P<0.01);实验组不同时间点之间进行比较,差异有统计学意义(F=278.62,P<0.01),且各时间点之间两两比较差异均有统计学意义(P<0.05)。
2.3 VDR在心肌组织中的表达抗体说明书标明现已明确有多量VDR表达的组织为小肠黏膜组织,所以本实验取得BALB/c小鼠小肠黏膜组织作为阳性切片,用来和心肌组织对比,同时用小肠黏膜组织协助摸索抗体最佳稀释浓度。镜下可见小肠黏膜组织VDR有较多表达;以PBS替代一抗的阴性对照切片未见棕色颗粒表达;正常对照组小鼠有极少量阳性表达。实验组小鼠第3、7、14和28天均有阳性表达,棕色颗粒主要表达于心肌细胞,染色加深,血管内皮细胞、炎症细胞也有少量表达。镜下观察发现,在第3天,VDR有少量表达;第7天时阳性表达明显增多,染色加深;第14天,VDR表达有所下降,但仍可见到较多表达;第28天,VDR表达明显下降。结果如图 2所示。
 |
图 2 免疫组织化学染色观察小鼠心肌以及小肠组织的 VDR 表达情况(×400) A:正常对照组可见极少量阳性表达;B:实验组第 3 天,可见少量阳性表达;C:实验组第 7 天,阳性表达明显增多;D:实验组第 14 天,可见较多阳性表达;E:实验组第 28 天,可见少量阳性表达;F:阴性对照,未见阳性表达; G:小肠黏膜阳性对照,可见较多阳性表达。图中阳性表达呈深棕色。 |
据统计分析,在第3、7、14和28天,实验组小鼠心肌VDR表达量相比正常对照组小鼠明显升高,差异均有显著统计学意义(P<0.01);对不同时间段实验组VDR水平进行比较,差异有统计学意义(P<0.01),进一步将各时间点数据行两两比较发现,第7天表达最高,第14天稍有下降,第3天和第28天表达最少,差异有统计学意义(P<0.05)。见表 1。
| 表 1 实验组与对照组小鼠心肌 VDR 表达水平比较(x±s) |
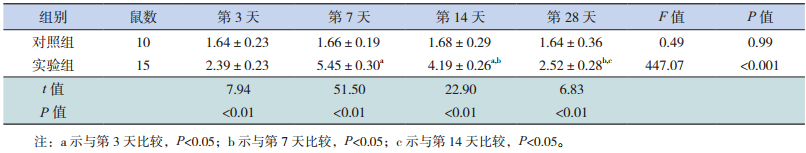
将实验组小鼠第3、7、14和28天VDR表达量与其病理组织变化情况进行相关性分析,结果表明实验第3、7、14和28天VMC小鼠心肌VDR的表达量与病理积分均存在较好的正性线性相关(分别r=0.927、0.894、0.820、0.859,均P<0.01)。
3 讨论VMC是由嗜心肌病毒引起的心肌细胞、心肌间质血管成分以及心包膜的炎性免疫反应性疾病。引起VMC的病原体有腺病毒、肠道病毒、埃可病毒和肝炎病毒等,其中肠道病毒中的CVB3作为VMC的最主要病原体受到越来越多的关注[9],CVB 3可以通过柯萨奇-腺病毒受体的介导感染损伤心肌[10],且VMC的易感性存在性别差异[11]。VMC的致病机制主要有病毒的直接侵害和由心肌损伤引发的免疫病损,最终导致心肌坏死和纤维化。
越来越多的研究揭示VMC存在过度免疫和免疫失衡:如TNF和IL-1等炎症因子在VMC病程中的过度表达与心肌免疫损伤存在密切关联[12, 13],Th1与Th2的失衡也参与了VMC免疫损伤,此外,Th17通过分泌IL17参与VMC的免疫调控[14],Th17与调节性T细胞的平衡是VMC免疫损伤研究的重要新靶点。
经研究发现VDR与其配体维生素D不仅参与钙磷代谢调节,且与各种免疫炎性疾病有着很大的关联,如哮喘、类风湿关节炎、炎性肠病和糖尿病等。在本实验中,BALB/c小鼠腹腔接种CVB3后,心肌的VDR表达水平明显增多。第3天,实验组小鼠心肌的VDR表达量相比对照组可见增多,此时MVC小鼠心肌细胞排列紊乱,有少量炎症细胞浸润聚集和心肌点状坏死;在第7天,实验组小鼠心肌的VDR表达水平达到高峰,此时MVC小鼠心肌可见大量炎症细胞浸润以及大片坏死病灶;到第14天,VDR表达仍然保持着较高水平,MVC小鼠心肌组织仍可见到多量炎性细胞浸润和片状坏死;第28天时,心肌组织炎症坏死大部分已吸收,VDR表达水平降低,但仍然高于正常对照组。进一步分析发现,VDR在MVC小鼠心肌的表达水平与心肌炎症坏死程度呈正相关,推测VDR参与了MVC炎性免疫病程。VMC小鼠心肌炎症坏死最明显时,其心肌VDR表达最多,恢复期则下降,推究VDR在VMC小鼠心肌中表达上调的可能机制是:病原入侵时,活化的淋巴细胞、巨噬细胞以及树突状细胞能促进维生素D转变为体内生物活性最强的1,25-二羟维生素D3,此活化作用受到诸多炎性介质的调控,同时活化的巨噬细胞又能自分泌1,25-二羟维生素D3,而高活性的1,25-二羟维生素D3能激活VDR的调控基因,促进了VDR的表达[4, 15]。足量的VDR与1,25-二羟维生素D3结合后,参与MVC炎性免疫病程。
在对其他同样有着炎性免疫病程疾病的研究中,Wittke A研究发现,在敲除VDR基因的哮喘小鼠不能形成炎症反应并缺乏气管高反应性,提示VDR缺失对Th2直接关联的免疫炎性疾病有保护作用[16]。同时,越来越多研究发现,补充足够量的维生素D能够抑制诸多免疫炎性性疾病病损,有助于炎性疾病的预后[17],而维生素D发挥生理效应与足够的VDR表达密切相关;更有学者发现在缺乏VDR表达的炎性肠病中,肠道病损更加严重[18],提示足够的VDR表达对炎性免疫疾病有保护作用;此外研究还发现,VDR受体激活剂能够改善心血管疾病终末期心肌纤维化和保护心肌舒张功能[19]。Boonstra A等人认为维生素D与VDR结合后,能通过抗原呈递细胞抑制Th1主要参与的免疫损害,从而起到减小此类免疫性疾病的免疫病损的作用[20],而Th1主要介导的是细胞免疫反应,此类免疫反应主要包括宿主对肿瘤或胞内病原体如病毒的免疫反应,当Th1攻击宿主自身成分时亦可引起自身免疫疾病。已知VMC是由病毒攻击心肌组织引起的疾病,且Fuse K等人经研究已指出,在VMC的病损期Th1参与的免疫反应占主要优势[21]。
小儿VMC能导致心律失常、阿斯综合征、充血性心衰、以及扩张性心肌病,如何积极有效的预防以及治疗VMC成为临床亟待解决的难题。本实验发现,VDR表达量与心肌病损程度有着密切的关系,相信可以通过进一步研究VDR和VMC发病之间的关系,进而为VMC的防治方法注入新的元素。
| [1] | Hendrickson WK, Flavin R, Kasperzyk JL, et al. Vitamin D receptor protein expression in tumor tissue and prostate cancer progression[J]. J Clin Oncol, 2011, 29(17):2378-2385. |
| [2] | Xiang W, Kong J, Chen S, et al. Cardiac hypertrophy in vitamin D receptor knockout mice:role of the systemic and cardiac renin-angiotensin systems[J]. Am J Physiol Endocrinol Metab, 2005, 288(1):E125-E132. |
| [3] | Girgis CM, Clifton-Bligh RJ, Hamrick MW, et al. The roles of vitamin D in skeletal muscle:form, function, and metabolism[J].Endocr Rev, 2013, 34(1):33-83. |
| [4] | Basson A. Vitamin D and Crohn's disease in the adult patient:a review[J]. JPEN J Parenter Enteral Nutr, 2014, 38(4):438-458. |
| [5] | 郭桂梅, 王娟, 夏敏, 等. 过敏性紫癜患儿血浆 1, 25(OH) 2D3, 维生素 D 受体和 24-羟化酶表达的意义[J]. 中华实用儿科临 床杂志, 2013, 28(21):1640-1642. |
| [6] | Guillot X, Semerano L, Saidenberg-Kermanac'h N, et al. Vitamin D and inflammation[J]. Joint Bone Spine, 2010, 77(6):552-557. |
| [7] | Kahlert C, Bergmann F, Beck J, et al. Low expression of aldehyde deyhdrogenase 1A1 (ALDH1A1) is a prognostic marker for poor survival in pancreatic cancer[J]. BMC Cancer, 2011, 11(1):275-284. |
| [8] | Guo CY, Han B, Chang H, et al. Anti-perforin neutralizing antibody reduces myocardial injury in viral myocarditis[J]. Cardiol Young, 2009, 19(6):601-607. |
| [9] | Jeserich M, Brunner E, Kandolf R, et al. Diagnosis of viral myocarditis by cardiac magnetic resonance and viral genome detection in peripheral blood[J]. Int J Cardiovasc Imaging, 2013, 29(1):121-129. |
| [10] | Yuen S, Smith J, Caruso L, et al. The coxsackie-adenovirus receptor induces an inflammatory cardiomyopathy independent of viral infection[J]. J Mol Cell Cardiol, 2011, 50(5):826-840. |
| [11] | Li K, Xu W, Guo Q, et al. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis[J]. Circ Res, 2009, 105(4):353-364. |
| [12] | Huang CH, Vallejo JG, Kollias G, et al. Role of the innate immune system in acute viral myocarditis[J]. Basic Res Cardiol, 2009, 104(3):228-237. |
| [13] | Fairweather D, Frisancho-Kiss S, Njoku DB, et al. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL-1β, and immune complex deposition in the heart[J]. J Immunol, 2006, 176(6):3516-3524. |
| [14] | Yuan J, Yu M, Lin QW, et al. Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis[J]. J Immunol, 2010, 185(7):4004-4010. |
| [15] | 朱慧花, 赵琳. 维生素D 与免疫功能的研究[J]. 医学综述, 2013, 19(5):820-822. |
| [16] | Wittke A, Weaver V, Mahon BD, et al. Vitamin D receptordeficient mice fail to develop experimental allergic asthma[J]. J Immunol, 2004, 173(5):3432-3436. |
| [17] | Querfeld U. Vitamin D and inflammation[J]. Pediatr Nephrol, 2013, 28(4):605-610. |
| [18] | Froicu M, Cantorna MT. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury[J]. BMC Immunol, 2007, 8(5):1-11. |
| [19] | Meems LM, Cannon MV, Mahmud H, et al. The vitamin D receptor activator paricalcitol prevents fibrosis and diastolic dysfunction in a murine model of pressure overload[J]. J Steroid Biochem Mol Biol, 2012, 132(3):282-289. |
| [20] | Boonstra A, Barrat FJ, Crain C, et al. 1α, 25-Dihydroxyvitamin D3 has a direct effect on naive CD4+ T cells to enhance the development of Th2 cells[J]. J Immunol, 2001, 167(9):4974-4980. |
| [21] | Fuse K, Kodama M, Aizawa Y, et al. Th1/Th2 balance alteration in the clinical course of a patient with acute viral myocarditis[J]. Jpn Circ J, 2001, 65(12):1082-1084. |