 2015, Vol. 17
2015, Vol. 17



2. 南华大学第二附属医院, 湖南 衡阳 421000
血友病A(Hemophilia A,HA) 是一种X 连锁隐性遗传性凝血功能障碍性疾病,患病率为1/5 000~10 000 [1, 2, 3]。血浆中凝血因子Ⅷ(factor Ⅷ ,F Ⅷ)缺乏或者功能缺陷是该病的发病机制,临床表现为出血或出血倾向。根据临床症状及F Ⅷ凝结活性(factor Ⅷ clotting activity,F Ⅷ :C)可将血友病A 分为重型(F Ⅷ :C <1%)、中型(F Ⅷ :C1%~5%)和轻型(F Ⅷ :C >5%~25%)。轻型血友病A患者多表现为自发或外伤后皮下出血及鼻衄,中、重型患者会有关节出血甚或重要脏器出血而危及生命。该病目前尚无有效根治措施,因此采用直接或间接基因诊断[4],准确检测出携带者、进行产前诊断可阻断致病基因传递,是防止患儿出生、降低本病发病率的主要措施。本研究就7 个血友病A 家系进行基因类型及临床表型分析,初步探讨血友病A 的分子机制,以期应用于血友病A 携带者诊断及产前诊断。 1 资料与方法 1.1 研究对象
选取2009 年10 月至2012 年10 月于广西医科大学第一附属医院确诊为血友病A 的8 例患者及其家系为研究对象,8 例血友病A 患者来自于7 个家系,均为男性,年龄范围3~11 岁,所有患者的诊断均符合张之南等[5] 主编的《血液病学》,10 名家系成员(女)肝功能均正常,近期未曾使用抗凝剂。 1.2 标本采集
在获得所有患者及其家系成员的知情同意书后,抽取外周静脉血4 mL,其中2 mL 用于DNA提取,另2 mL 送广西医科大学第一附属医院检验科用于F Ⅷ :C 和凝血功能的测定。 1.3 引物设计
参考文献[6, 7] 设计引物用于内含子22、内含子1 倒位检测,参考文献[8, 9] 合成引物28 对,覆盖了F Ⅷ基因所有外显子及启动子序列,用于F Ⅷ基因测序。以上引物均由上海生工生物工程技术公司合成。 1.4 内含子倒位检测
采用长链PCR(long distance PCR,LD-PCR)法及双管双重PCR 法,分别对所有血友病A 患者进行F Ⅷ内含子22 倒位及内含子1 倒位检测,检测阳性者进行相应家系检测。 1.5 F Ⅷ基因测序
采用DNA 直接测序法对内含子22 和内含子1 倒位检测阴性的患者进行F Ⅷ基因突变检测。通过chromas 软件分析测序图,与已知的F Ⅷ基因序列NG_011403.1 比对得出F Ⅷ基因突变类型,查询F Ⅷ 结构和突变位点数据库(HaemophiliaA Mutation,Structure,Test and Resource Site,HAMSTeRS)(http://hadb.org.uk/WebPages/Main/intrquiz.htm)、人类基因突变数据库(http://www.hgmd.cf.ac.uk/docs/new_back.html)以及美国国立生物技术信息中心(http://www.ncbi.nlm.nih.gov/)进一步了解是否为新突变,检出新突变位点后进行反向测序证实,并以50 例正常男性作为对照除外基因多态性。 2 结果 2.1 血友病A 患者临床资料分析
患者临床分型及常见出血部位见表 1,8 例患者中,重型2 例,就诊时表现为皮肤、黏膜等部位的轻微出血;中型6 例,其中5 例有关节出血,1 例仅有皮肤瘀斑。
| 表 1 8 例血友病A 患者F Ⅷ基因型、临床分型及出血表现 |
本研究未检测出内含子22 倒位患者,仅检测到1 例内含子1 倒位患者,对其家系成员进行相应检测,发现其母亲及姨妈均为内含子1 倒位携带者,结果见图 1。
 |
图 1 双管双重PCR 法检测内含子1 倒位电泳图 泳道2、6 分别为8 号患者Int1h-1、Int1h-2 电泳结果,显示条带为1.3 kb 和1.7 kb,为F Ⅷ内含子1 倒位;泳道3、7 为其母亲Int1h-1、Int1h-2 电泳结果,显示条带为1.3 kb、1.9 kb 和1.1 kb、1.7 kb,为F Ⅷ内含子1 倒位携带者;泳道4、8 为其姨妈Int1h-1、Int1h-2 电泳结果,同样为内含子1 倒位携带者;泳道1、5 为阴性对照;泳道9、10 为空白对照。 |
经测序筛检出基因突变患儿7 例,分别位于F Ⅷ基因外显子14 和23,其中6 例突变位于外显子14,1 例位于外显子23。检出5 种基因型,均为单碱基变异,其中错义突变2 例(c.2161A>G),缺失突变3 例(c.4379delA 和c.3666delA),插入突变1 例(c.4379_4380dupA),无义突变1 例(c.6496 C>T)(表 1)。患儿6 检出基因型p.His1202LeufsX16(c.3666delA),其母亲为杂合子(图 2),该基因型在HAMSTeRS、人类基因突变数据库以及美国国立生物技术信息中心等数据库中均未能检索到,50 例正常男性中亦未检测到相同基因变异,NCBI 数据库未检索到多态性基因记录,提示为新突变类型。
 |
图 2 患儿6 基因测序结果患儿为c.3666delA 纯合突变;患儿母亲为 c.3666delA 杂合突变。 |
7 家系中,共有10 名女性家系成员接受携带者检测,其中有5 名女性家系成员携带血友病A基因。见图 3。
 |
图 3 7 个血友病A 家系图 1~8 为患者编号□为健
康男性;○为健康女性; 为死亡男性; 为死亡男性;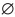 为死亡女性;■为男性
患者; 为死亡女性;■为男性
患者; 为女性携带者;箭头所指为先证者。 为女性携带者;箭头所指为先证者。
|
F Ⅷ基因定位于染色体Xq28,全长186 kb,由26 个外显子和25 个内含子组成,编码包含2 332 个氨基酸残基的多肽链,以A1-A2-B-A3-C1-C2 的顺序排列,A 区与F Ⅷ的蛋白质合成及其活化密切相关,B 区与F Ⅷ发挥活性无关,但能够调节F Ⅷ的分泌,该区单碱基缺失或插入会引起读码框移,进而影响正常剪切或转录的进行。F Ⅷ基因结构庞大,突变种类繁多,存在着高度异质性,目前已发现2 517 种基因突变注册在HGMD网站,包括错义/ 无义突变、缺失、插入突变和重复突变等,其中近60% 为错义/ 无义突变,重复突变在国内未见相关报道。血友病A 中重型患者大约45% 为F Ⅷ内含子22 倒位,3%~5% 为F Ⅷ内含子1 倒位[10],而在中型或轻型患者当中多见的是新发和散发的点突变、小的插入或缺失等[11]。
F Ⅷ内含子22 或内含子1 倒位多致重型血友病A[12]。本研究8 例患者通过LD-PCR 均未检测出内含子22 倒位,可能与检测样本数较少有关。患者8 临床分型为重型,经双管多重PCR 检出内含子1 倒位,其母亲及姨妈均为倒位携带者。
目前认为,外显子14 是F Ⅷ基因突变热点区域,截至2013 年3 月HAMSTeRS 数据显示,发生于F Ⅷ外显子14 的突变类型多达395 种,为总突变类型的18.7%,由于多数突变位于外显子14,目前该外显子已成为 F Ⅷ基因突变研究的热点[13]。本研究发现5 种基因型,有4 种位于外显子14。7 个家系中,有3 个家系患者突变位于外显子14 的一个A8 区域(c.4372-4379):A8 区域的单碱基A 插入或缺失均使得终止密码TAA 或TAG 提前出现,导致F Ⅷ发生截断突变。既往研究认为,F Ⅷ基因复制过程中,在单碱基连续的区域容易发生滑动,因而导致插入/ 缺失事件发生,其中A 连续的区域最多见,多数表型为重型[14]。本文3 例突变发生于A8 区域的患者均为散发病例,临床表型为中型或重型,多有关节出血等严重出血表现,与文献一致。患者1、2 为同一家系,基因型p.Met721Val,基因变异发生于B 区域,两者F Ⅷ :C 均小于2%,临床上均有关节出血表现,推测其基因突变可能影响到复合物构象结构的稳定,既往曾有报道,但致病机制尚未十分明确[15]。
患者6 突变位于B 区,基因型p.His1202LeufsX16,其母亲为杂合子,该基因型在HAMSTeRS、HGMD 及NCBI 等数据库中未能检索到。对50 名正常男性在相应碱基位置进行直接测序均未发现同样碱基改变,提示该突变为新突变类型。推测单碱基A 的缺失导致框移突变,终止密码提前出现,最终造成F Ⅷ的截断,从而影响F Ⅷ的功能。
患者5 的F Ⅷ基因突变位于外显子23,基因型p.Arg2166X,发生在C1 区的无义突变将导致表达产物F Ⅷ缺少C2 区,将严重影响F Ⅷ与vWF的结合,使F Ⅷ在体内不能正常循环代谢[16]。受累患者为3 岁男孩,临床诊断为中型,并出现关节出血症状。
7 个家系中,有4 个家系共5 名女性成员携带血友病A 基因(图 3),其余散发病例的F Ⅷ基因变异均发生于多聚A序列的单碱基插入或缺失,显示多聚A 在复制过程中容易发生滑动而导致基因突变,且多为非遗传性。
既往认为,血友病A 患者的F Ⅷ基因型与临床表型有着较好的相关性,通过明确基因型可以预测患者的临床出血程度,但是研究发现,这种相关性并不多见,仅体现于大片段缺失、无义突变等基因型[17]。本研究发现无义突变1 例,为中型,表现为皮肤、关节出血;错义突变2 例,均为中型,伴有关节出血;框移突变后发生F Ⅷ蛋白截断1 例,亦为中型,亦有关节出血。值得一提的是,患者7 基因型为p.Asn14061LysfsX1,患者8为内含子1 倒位,临床均归为重型,但仅有皮肤、黏膜等较为轻微的出血表现;可见F Ⅷ :C 或F Ⅷ基因型并不是决定患者临床表型的唯一因素,可能还存在其他影响F Ⅷ功能的调节机制。新基因型p.His1202LeufsX16,其患者临床分型为中型,但有关节出血等较严重的出血表现,该基因变异对于F Ⅷ凝血功能的影响机制值得进一步探究。
| [1] | 中华医学会血液学会分会血栓与止血学组, 中国血友病协作组 . 血友病诊断与治疗中国专家共识 (2013 版 ) [J]. 中华血液学杂志, 2013, 34(5): 461-463. |
| [2] | Keeney S, Mitchell M, Goodeve A. The molecular analysisof haemophilia A: a guideline from the UK haemophiliacentre doctors, organization haemophilia genetics laboratorynetwork[J]. Haemophilia, 2005, 11(4): 387-397. |
| [3] | 梁燕, 赵耘, 王战勇, 等. 血友病甲基因分析技术的改进及其在产前诊断中的应用 [J]. 中华医学遗传学杂志, 2007, 24(4):437-439. |
| [4] | 周竣荔, 韦红英, 吴华, 等. STR 遗传标记在广西地区血友病A 携带者诊断中的运用 [J]. 中国当代儿科杂志, 2012, 14(14):951-955. |
| [5] | 张之南, 杨天楹, 郝玉书. 血液病学 [M]. 北京 : 人民卫生出版社, 2003: 1724-1725. |
| [6] | Liu Q, Nozari G, Sommer SS. Single-tube polymerase chainreaction for rapid diagnosis of the inversion hotspot of mutationin hemophilia A[J]. Blood, 1998, 92(4): 1458-1459. |
| [7] | 李坦, 戴菁, 吴竞生, 等. 血友病 A 患者凝血因子Ⅷ基因内含子 1 及 22 倒位分析 [J]. 中华血液学杂志, 2009, 30(3): 150-153. |
| [8] | Viel KR, Machiah DK, Warren DM, et al. A sequence variationscan of the coagulation factor VIII (FVIII) structural gene andassociations with plasma FVIII activity levels[J]. Blood, 2007,109(9): 3713-3724. |
| [9] | Lin SY, Su YN, Hung CC, et al. Mutation spectrum of 122hemophilia A families from Taiwanese population by LD-PCR,DHPLC, multiplex PCR and evaluating the clinical applicationof HRM[J]. BMC Med Genet, 2008, 9: 53. |
| [10] | Bagnall RD, Waseem N, Green PM, et al. Recurrent inversionbreaking intron l of the factor VIII gene is a frequent cause ofsevere hemophilia A[J]. Blood, 2002, 99(1): 168-174. |
| [11] | Nadja B, Ameni M, Reswith E, et al. Spectmm of moleculardefects and mutation deteetion rate in patients with mild andmoderate hemophilia A[J]. Hum Mutat, 2007, 28(1): 54-60. |
| [12] | Zimmermann MA, Oldenburg J, Müller CR, et al. Unusualgenomic rearrangements in introns 1 and 22 of the F8 gene[J].Hamostaseologie, 2011, 31 Suppl 1: S69-S73. |
| [13] | Bogdanova N, Markoff A, Pollmann H, et al. Prevalence ofsmall rearrangements in the factor VIII gene F8C amongpatients with severe hemophilia A[J]. Hum Mutat, 2002, 20(3):236-237. |
| [14] | Pelak K, Shianna KV, Ge D, et al. The characterization oftwenty sequenced human genomes[J]. PLoS Genet, 2010, 6(9):e1001111. |
| [15] | Santacroce R, Acquila M, Belvini D, et al. Identification of217 unreported mutations in the F8 gene in a group of 1,410unselected Italian patients with hemophilia A[J]. J Hum Genet,2008, 53(3): 275-284. |
| [16] | 李汶, 胡晓, 高伯笛, 等. 10 个甲型血友病家系 F Ⅷ基因突变分析 [J]. 中华遗传学杂志, 2011, 28(2): 127-132. |
| [17] | Bowen DJ. Haemophilia A and haemophilia B: molecularinsights[J]. Mol Pathol, 2002, 55(1): 1-18. |