 2016, Vol. 18
2016, Vol. 18


中、短链酰基辅酶A脱氢酶缺乏症(medium chain acyl-CoA dehydrogenase deficiency,MCADD,OMIM 201450; short chain acyl-CoA dehydrogenase deficiency,SCADD,OMIM 201470)是由于基因突变致使相应的酰基辅酶A脱氢酶功能发生缺陷,引起中、短链脂肪酸β氧化障碍,从而导致能量生成减少和代谢中间产物在体内大量蓄积,属常染色体隐性遗传代谢性疾病[1-2]。该病主要以新生儿期发病多见,临床表现为低血糖及酸中毒反复发作难以纠正、癫癎、肌张力低下、发育迟缓以及精神发育迟滞等。我们对2例顽固性低血糖合并代谢性酸中毒的患儿进行了血液酯酰肉碱谱和尿液有机酸分析,结果高度疑似MCADD和SCADD,进一步对家系成员进行相关基因突变检测(包括ACADM及ACADS基因),为患儿的治疗和随访以及今后的产前诊断提供理论依据。
1 资料与方法 1.1 研究对象家系1:患儿,男,3 d,第1胎第1产,足月顺产出生,出生体重3 000 g。因新生儿窒息、反应差、吸奶无力、嗜睡在我院新生儿科治疗。体格检查:体温36.4℃,心率153 次/min,呼吸45 次/min,血压72/41 mm Hg,体重3 020 g,面容正常,面色红润,前囟张力稍高,双瞳孔等大等圆,对光反射存在,颈软,双肺呼吸音粗,心音有力,律齐,心前区未闻及杂音,腹平软,未触及异常包块,肝脾肋下未触及,质中,四肢肌张力稍增高,脑膜刺激征阴性。血气分析:pH 7.22,PCO2 44 mm Hg,PO2 42 mm Hg,BE -9.61 mmol/L,HCO3 17.8 mmol/L,提示代谢性酸中毒;血糖1.5 mmol/L(参考值:2.6~7.0 mmol/L);尿酮体阴性;血氨、肝肾功能电解质等检查均未见明显异常。患儿父母双方体健,非近亲结婚,否认家族遗传疾病史。
家系2:患儿,女,3个月,第1胎第1产,足月顺产出生,出生体重3 200 g,Apgar评分10分,因咳嗽伴发热10余天在我院重症医学科治疗。体格检查:体温37.9℃,心率142 次/min,呼吸34次/min,血压78/44 mm Hg,体重3.3 kg,神清,精神稍差,哭时泪少,皮肤、口唇干燥,皮肤弹性欠佳,双瞳孔等大等圆,对光反射灵敏,口周无发绀,口腔黏膜光滑,呼吸尚平稳,未见吸气性三凹征,咽充血,双肺呼吸音稍粗,可闻及少许固定细湿罗音,心音稍低钝,律齐,心前区未闻及杂音,腹平软,肝脾肋下未触及,神经系统检查未见异常。实验室检查:多次血糖检测结果均低于2.6 mmol/L(参考值:2.6~7.0 mmol/L);血气分析:pH 7.41,PO2 108 mm Hg,PCO2 23 mm Hg,BE -8.0 mmol/L,HCO3 14.6 mmol/L,提示代谢性酸中毒;电解质:K+ 1.5 mmol/L(参考值:3.5~5.5 mmol/L),Na+ 130 mmol/L(参考值:135~145 mmol/L),余检查未见明显异常。母亲孕期血糖检查正常,无糖尿病史,否认家族遗传病史。
1.2 血液酯酰肉碱谱及尿液有机酸分析采集患儿末梢血滴于滤纸片(英国沃特曼公司,S&S903#)上,室温自然晾干后,将血滤纸片打孔置于96孔过滤板中,每孔加入含氨基酸和酰基肉碱同位素内标的甲醇300 μL,室温密封震荡30 min,萃取血片中的氨基酸和酰基肉碱,然后离心至另一个96孔聚丙烯板,50℃加热氮气吹干,再加入50 μL盐酸正丁醇(3 mol/L),Teflon膜覆盖,置65℃恒温箱内15 min,随后50℃氮气吹干,再加入80%乙腈100 μL溶解,铝膜覆盖后上样检测。根据同位素内标和各种丁酯化的氨基酸和酰基肉碱的离子峰强度,采用定量分析软件,由已知浓度的内标自动计算出所测样品中氨基酸和酰基肉碱的浓度。
同时随机留取尿于滤纸片(英国沃特曼公司,S&S903#)上,干燥后送武汉康圣环球医学检验所进行尿有机酸分析。
1.3 ACADM及ACADS基因突变分析根据知情同意原则,抽取患儿及父母外周静脉血2 mL,EDTA抗凝处理,常规酚-氯法提取基因组DNA。针对ACADM、ACADS基因外显子,采用Primer Premier 5.0设计引物,扩增全部外显子以及与外显子交界的部分内含子区域。PCR反应体系为25 μL,包括TaKaRa LA Taq premix 12.5 μL,上、下游引物混合液0.75 μL(10 pmol/μL),基因组DNA100 ng,加去离子水至25 μL。PCR反应条件:95℃预变性3 min;94℃变性30 s,60℃退火30 s,72℃延伸40 s,38个循环;72℃延伸8 min。PCR产物经1.5%琼脂糖凝胶电泳鉴定。PCR扩增产物送英骏生物公司测序,结果与人类基因组ACADM、ACADS基因序列进行比较。
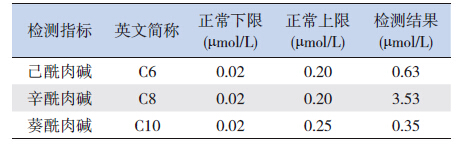
2 结果 2.1 血液酯酰肉碱谱及尿液有机酸分析结果家系1患儿中链酰基肉碱增高(表 1),其中辛酰肉碱(C8)3.52 μmol/L(正常参考值0.02~0.2 μmol/L)(图 1),尿有机酸分析未见明显异常;家系2患儿血丁酰肉碱(C4)1.66 μmol/L(正常参考值0.06~0.6 μmol/L)(图 2),尿有机酸分析示乙基丙二酸为55.9(正常参考值0~6.2)。
| 表 1 家系1 患儿中链酰基肉碱检测结果 |

|
图 1 家系1 患儿血液酯酰肉碱谱分析结果 辛酰肉碱(C8)显著增高。 |

|
图 2 家系2 患儿血液酯酰肉碱谱分析结果 丁酰肉碱(C4)显著增高。 |
2.2 ACADM、ACADS基因测序结果
Sanger测序发现家系1患儿ACADM基因已报道纯合突变c.580A>G(p.Asn194Asp)[3],使得ACADM基因编码氨基酸第194位天冬酰胺被天冬氨酸替代(图 3);家系2患儿ACADS基因已报道纯合突变c.625G>A(p.Gly209Ser)[4],使得ACADS基因编码氨基酸第209位甘氨酸被丝氨酸替代。2例患儿突变均来自父母双方(图 4)。

|
图 3 家系1 患儿ACADM 基因测序结果 ①正常对 照序列;②患儿ACADM 基因7 号外显子c.580A>G(p.Asn194Asp) 纯合突变(箭头所指);③④分别为父母ACADM 基因7 号外显 子c.580A>G(p.Asn194Asp)杂合突变(箭头所指)。 |

|
图 4 家系2 患儿ACADS 基因测序结果 ①正常对 照序列;②患儿ACADS 基因6 号外显子c.625G>A(p.Gly209Ser) 纯合突变(箭头所指);③④分别为父母ACADS 基因6 号外显子 c.625G>A(p.Gly209Ser)杂合突变(箭头所指)。 |
2.3 治疗经过及随访
家系1患儿在发病初期确诊MCADD即给予升糖及补液纠正酸中毒等治疗,同时予左卡尼汀每日100 mg/kg口服,低血糖和酸中毒较前好转。现患儿门诊定期随访观察治疗,生长发育正常,复查血酰基肉碱C8仍偏低,尿有机酸未见异常。家系2患儿现予核黄素每日10 mg/kg及左卡尼汀每日100 mg/kg口服治疗,病情平稳。门诊定期复查随访,运动发育较正常同龄儿童稍落后,复查血酯酰肉碱提示丁酰肉碱仍偏低,但尿液乙基丙二酸水平恢复正常。
3 讨论脂肪酸是机体在应激状态下通过线粒体β氧化为机体供能的主要物质来源,其过程需要多种酶催化完成。近年来研究发现在脂肪酸β氧化过程中大约有20余种活性酶参与,其中中链酰基辅酶A脱氢酶(medium chain acyl-CoA dehydrogenase,MCAD)和短链酰基辅酶A脱氢酶(short-chain acyl-CoA dehydrogenase,SCAD)是脂酰基辅酶A脱氢酶家族中的一员,特异性分解相应的底物,是脂肪酸β氧化脱氢进行电子传递的限速酶[5]。MCAD和SCAD的结构具有高度同源性,位于线粒体基质中,在肝脏、骨骼肌、心肌、皮肤成纤维细胞中均有表达,如其结构或功能缺陷或活性下降,则会引起神经系统、骨骼肌、心、肝、肾等重要器官功能异常,在临床上常表现为低血糖、心肌病、肌张力低下、酸中毒以及脑病等症状[6]。除了本文所提及的MCADD和SCADD之外,脂肪酸β氧化代谢障碍还包括肉碱转运障碍、肉碱棕榈酰转移酶缺乏症、以及长、极长链酰基辅酶A脱氢酶缺乏症等十余种遗传代谢性疾病[7]。
MCAD前体蛋白由421个氨基酸构成,其中前25个氨基酸为前导肽,剩余的氨基酸为成熟多肽,经折叠后形成同源四聚体与电子转运黄素蛋白相互作用。其编码基因ACADM位于常染色体1p31.1,包含12个外显子,目前已报道95种突变,以错义突变为主要突变类型,约占突变总类型的60%。其中最常见的突变是位于11号外显子的c.985A>G,导致第304位赖氨酸被谷氨酸取代。Khalid等[8]研究发现,K304E纯合突变的患儿种族94%为白种人,亚裔和黑色人种当中鲜有报道,提示该突变有种族特异性,估算此突变的携带率约为1/65。亚裔日本人群中449_452del4突变最为常见,目前我国尚无关于MCADD患者突变谱及突变热点的报道。本例患儿通过基因检测确诊,提示为7号外显子纯合突变c.580A>G(p.Asn194Asp),使得ACADM基因编码氨基酸第194位天冬酰胺被天冬氨酸替代,导致MCAD无效表达,严重影响其催化性能及特异性,降低了与底物的亲和力[9]。目前,MCADD基因型与临床表型的关系尚不明确,由于基因与基因的相互作用,基因与环境的相互作用,均可影响该病的自然进程,所以通过基因型不能准确预测患者的临床表现及疾病的严重程度[10]。
MCADD属罕见的脂肪酸代谢疾病,于1983年由Rhead等首次报道。近年来,随着新生儿串联质谱筛查的开展,MCADD发病率逐渐明确。不同国家和地区差异较大,其中白种人患病率较高,英国约9.4/10万,加拿大约7.1/10万,澳大利亚约5.2/10万,美国约5.2/10万,德国约12.3/10万,荷兰约21.1/10万,奥地利约4/10万,沙特阿拉伯约5.5/10万,此病在北欧地区是继苯丙酮尿症后第二位常见的遗传代谢病[11-14]。亚洲发病率较低,日本新生儿发病率约1.9/10万,上海新华医院筛查54万新生儿确诊4例,患病率约0.7/10万[15]。本病多在出生后3个月到3岁之间发病,少数在新生儿期或成人期发病,也有无症状患者。发病通常都有诱发因素存在,以长时间饥饿最为常见,并发感染也是常见原因。本病临床表现多样[16],早期发病患儿,首发症状以嗜睡和呕吐常见,也可表现为抽搐、窒息等,常迅速进展为昏迷或死亡。姜涛等[17]报道1例MCADD患儿除有上述临床表现外还合并肝细胞损伤,患儿基因型c.572G>A(p.Trp191*)导致翻译终止。李甫棒等[18]报道1例以黄疸为首发症状的MCADD患儿同时合并代谢性酸中毒。王立娜等[19]报道1例以先心病合并肺炎起病的患儿,同时伴有代谢性酸中毒。急性发作期实验室检查常有低血糖表现,血糖降低伴尿酮体阴性有助于诊断,血酰基肉碱筛查可发现中链酰基肉碱升高,辛酰肉碱显著升高是该病的特征性变化。本例患儿发病初期有窒息、嗜睡等临床表现,同时患儿伴有肺炎感染诱发因素,实验室检查血糖偏低难以纠正,且存在明显的代谢性酸中毒,血酰基肉碱谱分析提示辛酰肉碱 3.52 μmol/L(正常参考值0.02~0.2μmol/L),符合MCADD的初步诊断。但该患儿尿有机酸分析未见异常,可能与尿标本留取时患儿已经度过急性发作期有关。MCADD无特异性临床表现,早期诊断主要依靠特殊的临床表现和实验室检查,其中血辛酰肉碱水平的筛查是MCADD最重要的特征性指标。但应当注意的是,当继发性肉碱缺乏时辛酰肉碱水平可能升高不明显,要结合C8/C10比值来提高诊断的敏感性和准确性。本病急性发作期尿气相色谱质谱有机酸分析可发现尿二羧酸浓度升高,但病情趋于稳定时可正常,因此尿有机酸分析只适用于病情发作时检测,通过酶学或基因检测有助于进一步明确诊断。
本病治疗总的原则是避免饥饿,急性发作期积极对症处理。Derks等[20]研究MCAD患儿可耐受饥饿时间最大值,推荐6个月~1岁为8 h,1~2岁为10 h,大于2岁可达12 h,婴幼儿期患儿需频繁喂养以提供充足热量摄入防止过度脂肪动员。急性发作期积极纠正低血糖和补充足量液体及电解质是改善代谢失衡和清除有毒代谢物的关键,对于血液游离肉碱显著降低的患者,可小剂量补充左卡尼汀。本例患儿在发病初期确诊MCADD即给予升糖及补液纠正酸中毒等治疗,同时予左卡尼汀每日100 mg/kg口服,低血糖和酸中毒较前好转。现患儿门诊定期随访观察治疗,生长发育正常,复查辛酰肉碱仍偏低,尿有机酸未见异常。
SCADD属常染色体隐性遗传代谢性疾病,其致病基因ACADS基因含10个外显子,编码412个氨基酸,至今已报道70余种基因突变类型,以错义突变为主。Gregersen等[21]及Jethva等[22]研究发现欧美国家和犹太人中以c.625G>A(G209S)和c.511C>T(R147W)突变为主。对美国694例新生儿筛查这两个突变位点,发现c.625G>A突变占所有检测等位基因的22%,c.511C>T占3%。另一项研究对荷兰1 036例新生儿筛查此位点发现5.5%为c.625G>A突变的纯合子,31.3%为c.625G>A突变的杂合子。SCADD在亚洲人群的发病率明显低于白种人,而c.625G>A突变在西班牙裔中携带率高达30%。这些突变在SCADD发病中的作用和机制尚不明确,但其在正常人群中也占有相当比例,提示它们不足以单独引起SCADD的发病,可能需要联合其他调控基因或环境因素共同参与方才导致SCAD酶活性降低和发病。在本研究中,SCADD患儿通过ACADS基因检测发现已知纯合突变c.625G>A(p.Gly209Ser),使得ACADS基因编码氨基酸第209位甘氨酸被丝氨酸替代,影响了SCAD结构和功能,导致脂肪酸β氧化无法正常进行。该结果与之前其他地区报道的突变热点相近,可以考虑将c.625G>A突变作为本地区该病常规基因筛查位点之一。
SCADD的发病率有种族和地区差异,研究表明,美国、德国、澳大利亚等国家新生儿疾病筛查提示其发病率约为1/95 000,目前我国对该病的发病率尚无明确统计。SCADD患者临床表现主要表现为神经系统方面,发育迟缓最为常见[23]。其他症状可有语言落后、肌张力低下、惊厥和行为问题,偶见急性酸中毒表现。Van Calcar等[24]对17例SCADD新生儿进行了出生后2年的随访研究,并未发现任何临床症状,但美国和澳大利亚新生儿疾病筛查研究发现SCADD患者临床表现多样,可伴有其他脂肪酸代谢障碍临床症状,如低血糖和酸中毒[25-26]。血丁酰基肉碱及尿液乙基丙二酸升高是SCADD诊断的主要参考指标,但乙基丙二酸升高并非SCADD的特异性改变,也可在戊二酸血症Ⅱ型和线粒体病中出现,应注意鉴别诊断,必要时进行基因诊断验证。本例患儿以新生儿期顽固性低血糖和代谢性酸中毒为主要临床表现,血丁酰肉碱1.66 μmol/L(正常参考值0.06~0.6 μmol/L)和尿液有机酸乙基丙二酸55.9(正常参考值0~6.2)显著增高,提示SCADD可能性大,通过ACADS基因检测发现已知纯合突变c.625G>A(p.Gly209Ser),使得ACADS基因编码氨基酸第209位甘氨酸被丝氨酸替代,影响了SCAD结构和功能,导致脂肪酸β氧化无法正常进行。
目前对SCADD的治疗尚无统一共识,主要处理措施是改善临床症状,低脂饮食,避免饥饿,可适当补充肉碱或维生素B2。对于急性发作期治疗与其他脂肪酸代谢障碍类似,可静脉给予10%的葡萄糖溶液抑制分解代谢。van Maldegem等[27]对核黄素(每日10 mg/kg)治疗16例患儿研究中发现,治疗后尿中乙基丙二酸排出减少,临床症状改善,停止摄入核黄素后症状复现,提示高剂量核黄素治疗SCADD可改善临床及生化指标。本例患儿门诊定期随访观察治疗,运动发育较正常同龄儿童稍落后,复查血酯酰肉碱提示丁酰肉碱仍偏低,但尿液乙基丙二酸水平恢复正常。
本研究通过血酰基肉碱谱及尿有机酸分析结合基因诊断的方法,对2例低血糖合并代谢性酸中毒患儿进行了从临床到分子的诊断,对遗传代谢病的治疗乃至预后都起到了指导作用。由于此类疾病致死致残率较高,早期识别、早期诊断、是本病预后的关键,目前我国部分地区已将此病列入新生儿疾病常规筛查项目,通过筛查酰基肉碱和尿有机酸,以及进一步基因检测即可确诊,避免了发生误诊和漏诊的可能性,使脂肪酸代谢障碍疾病的早期诊断成为可能。同时基因诊断为遗传咨询和产前诊断提供了重要信息,使患儿得到了及时的诊断和治疗,对于有再生育要求的家庭,建议进行产前诊断预防此类患儿出生。
| [1] | Rhead WJ, Amendt BA, Fritchman KS, et al. Dicarboxylic aciduria: deficient[J]. Science , 1983, 221 (4605) : 73–75. DOI:10.1126/science.6857268 |
| [2] | Amendt BA, Greene C, Sweetman L, et al. Short-chain acylcoenzyme A dehydrogenase deficiency. Clinical and biochemical studies in two patients[[J]. J Clin Invest , 1987, 79 (5) : 1303–1309. DOI:10.1172/JCI112953 |
| [3] | Nichols MJ, Saavedra-Matiz CA, Pass KA, et al. Novel mutations causing medium chain acyl-CoA dehydrogenase deficiency: under-representation of the common c.985 A>G mutation in the New York state population[J]. Am J Med Genet A , 2008, 146A (5) : 610–619. DOI:10.1002/(ISSN)1552-4833 |
| [4] | Corydon MJ, Gregersen N, Lehnert W, et al. Ethylmalonic aciduria is associated with an amino acid variant of short chain acyl-coenzyme A dehydrogenase[J]. Pediatr Res , 1996, 39 (6) : 1059–1066. DOI:10.1203/00006450-199606000-00021 |
| [5] | Finocchiaro G, Ito M, Tanaka K. Purification and properties of short-chain acyl-CoA, medium-chain acyl-CoA, and isovaleryl-CoA dehydrogenases from human liver[J]. Adv Neurol , 1988, 48 : 221–230. |
| [6] | Pedersen CB, Kølvraa S, Kølvraa A, et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level[J]. Hum Genet , 2008, 124 (1) : 43–56. DOI:10.1007/s00439-008-0521-9 |
| [7] | van Maldegem BT, Duran M, Wanders RJ, et al. Clinical, biochemical, and genetic heterogeneity in short-chain acylcoenzyme A dehydrogenase deficiency[J]. JAMA , 2006, 296 (8) : 943–952. DOI:10.1001/jama.296.8.943 |
| [8] | Khalid JM, Oerton J, Besley G, et al. Relationship of octanoylcarnitine concentrations to age at sampling in unaffected newborns screened for medium-chain acyl-CoA dehydrogenase deficiency[J]. Clin Chem , 2010, 56 (6) : 1015–1021. DOI:10.1373/clinchem.2010.143891 |
| [9] | Nichols MJ, Saavedra-Matiz CA, Pass KA, et al. ovel mutations causing medium chain acyl-CoA dehydrogenase deficiency: under-representation of the common c.985A>G mutation in the New York state population[J]. Am J Med Genet A , 2008, 146A (5) : 610–169. DOI:10.1002/(ISSN)1552-4833 |
| [10] | Gramer G, Haege G, Fang-Hoffmann J, et al. Medium-chain acyl-CoA dehydrogenase deficiency: evaluation of genotypephenotype correlation in patients detected by newborn screening[J]. JIMD Rep , 2015, 23 : 101–112. DOI:10.1007/978-3-662-47467-9 |
| [11] | Karaceper MD, Chakraborty P, Coyle D, et al. The health system impact of false positive newborn screening results for mediumchain acyl-CoA dehydrogenase deficiency: a cohort study[J]. Orphanet J Rare Dis , 2016, 11 : 12. DOI:10.1186/s13023-016-0391-5 |
| [12] | Andresen BS, Lund AM, Hougaard DM, et al. MCAD deficiency in Denmark[J]. Mol Genet Metab , 2012, 106 (2) : 175–188. DOI:10.1016/j.ymgme.2012.03.018 |
| [13] | Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation[J]. J Inherit Metab Dis , 2010, 33 (5) : 469–477. DOI:10.1007/s10545-010-9061-2 |
| [14] | Grünert SC, Wehrle A, Villavicencio-Lorini P, et al. Mediumchain acyl-CoA dehydrogenase deficiency associated with a novel splice mutation in the ACADM gene missed by newborn screening[J]. BMC Med Genet , 2015, 16 : 56. |
| [15] | 顾学范. 临床遗传代谢病[M]. 北京: 人民卫生出版社, 2015 : 146 -149. |
| [16] | 张惠文, 顾学范. 中链酰基辅酶A 脱氢酶缺乏症研究进展[J]. 国外医学(儿科学分册) , 2003, 30 (4) : 218–220. |
| [17] | 姜涛, 欧阳文献, 谭艳芳, 等. 中链酰基辅酶A 脱氢酶缺乏 症1 例肝脏病理分析及基因检测[J]. 临床儿科杂志 , 2016, 34 (4) : 249–252. |
| [18] | 李甫棒, 黄晓磊, 陈洁, 等. 以黄疸为首发症状中链酰基辅 酶A 脱氢酶缺乏症1 例报告[J]. 临床儿科杂志 , 2010, 28 (9) : 880–881. |
| [19] | 王立娜, 任常军, 李云, 等. 中链酰基辅酶A 脱氢酶缺乏症 合并先天性心脏病1 例[J]. 河北医药 , 2011, 33 (22) : 3520. |
| [20] | Derks TG, Touw CM, Ribas GS, et al. Experimental evidence for protein oxidative damage and altered antioxidant defense in patients with medium-chain acyl-CoA dehydrogenase deficiency[J]. J Inherit Metab Dis , 2014, 37 (5) : 783–789. DOI:10.1007/s10545-014-9700-0 |
| [21] | Gregersen N, Andresen BS, Pedersen CB, et al. Mitochondrial fatty acid oxidation defects-remaining challenges[J]. J Inherit Metab Dis , 2008, 31 (5) : 643–657. DOI:10.1007/s10545-008-0990-y |
| [22] | Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme A dehydrogenase deficiency[J]. Mol Genet Metab , 2008, 95 (4) : 195–200. DOI:10.1016/j.ymgme.2008.09.007 |
| [23] | Pedersen CB, Kølvraa S, Kølvraa A, et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level[J]. Hum Genet , 2008, 124 (1) : 43–56. DOI:10.1007/s00439-008-0521-9 |
| [24] | Van Calcar SC, Baker MW, Williams P, et al. Prevalence and mutation analysis of short/branched chain acyl-CoA dehydrogenase deficiency (SBCADD) detected on newborn screening in Wisconsin[J]. Mol Genet Metab , 2013, 110 (1-2) : 111–115. DOI:10.1016/j.ymgme.2013.03.021 |
| [25] | Bok LA, Vreken P, Wijburg FA, et al. Short-chain acyl-CoA dehydrogenase deficiency: studies in a large family adding to the complexity of the disorder[J]. Pediatrics , 2003, 112 (5) : 1152–1155. DOI:10.1542/peds.112.5.1152 |
| [26] | Jethva R, Ficicioglu C. Clinical outcomes of infants with shortchain acyl-coenzyme A dehydrogenase deficiency (SCADD) detected by newborn screening[J]. Mol Genet Metab , 2008, 95 (4) : 241–242. DOI:10.1016/j.ymgme.2008.09.003 |
| [27] | van Maldegem BT, Kloosterman SF, Janssen WJ, et al. High prevalence of short-chain acyl-CoA dehydrogenase deficiency in the Netherlands, but no association with epilepsy of unknown origin in childhood[J]. Neuropediatrics , 2011, 42 (1) : 13–17. DOI:10.1055/s-0031-1275342 |