 2016, Vol. 18
2016, Vol. 18



先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一组因肾上腺皮质激素合成途径中酶缺陷引起的疾病,属常染色体隐性遗传病,其中以21-羟化酶缺乏症(21-hydroxylase deficiency,21-OHD)最常见,占本病的90%~95% [1]。 21-OHD因不同的基因缺陷,可能对酶的影响不同导致临床表型的差异。根据酶缺乏的程度,可分为失盐型(salt wasting,SW)、单纯男性化型(simple virilizing,SV)和非经典型(non-classic,NC)3种。受累的酶21羟化酶由活性基因CYP21A2编码,该基因位于第6号染色体短臂上,与不具活性的假基因(CYP21A1P)串联排列组成。绝大多数突变源于有功能的CYP21A2基因和假基因之间的基因转换,假基因成了突变的源头[2]。已知突变120多种,约90%的突变是由9种点突变以及CYP21基因的缺失、转换引起[3, 4, 5]。据报道,CAH发病率有明显的种族和地区差别[6],但目前尚未有关新疆地区维吾尔族CAH的有关研究,因此本研究将有利于了解中国维吾尔族21-OHD基因突变规律及基因型和临床表型的关系。目前不同类型的21-OHD可通过临床症状和生化检测来诊断,但均可能发生误诊或漏诊,而对患者进行基因型分析,可以做到早期诊断和类型鉴别[7, 8]。在遗传咨询和产前诊断中,21-OHD的基因诊断是唯一可靠的方法[9]。
1 资料与方法 1.1 一般资料2013年10月至2014年10月新疆医科大学第一附属医院小儿内一科收治的18例21-OHD患儿因肾上腺危象或外生殖器畸形就诊,初诊年龄28 d至12岁;民族均为维吾尔族;父母均非近亲结婚;社会性别男8例,女10例,其中1例社会性别为男性者染色体性别为女性,其余社会性别均与染色体性别一致;12例为SV型,5例为SW型,1例为NC型。另2例于新生儿筛查时发现,其中男1例,女1例,均为染色体性别;民族均为维吾尔族;后经基因测序确诊,均为SW型。故本研究共纳入研究对象20例,临床表型中SV型12例,SW型7例,NC型1例;染色体性别男8例,女12例。
1.2 方法20例患儿除进行详细的体格检查外,均进行血17α-羟孕酮(17-OHP)、促肾上腺皮质激素(ACTH)、睾酮(T)、血钾、血钠、肾上腺B超及染色体核型分析。部分患儿检测血气分析、心电图、肾上腺CT及骨龄。其中T、雌二醇(E2)、卵泡生成素(FSH)、黄体生成素(LH)采用酶联免疫法测定;ACTH、皮质醇(COR)采用电化学发光法测定; 17-OHP采用放免法测定。
1.3 标本采集和DNA提取在知情同意的原则下,采集受检者外周血2 mL,置于EDTA抗凝管中。取EDTA抗凝外周血标本0.2 mL,按血液/细胞/组织基因组DNA提取试剂盒(北京天根生物)说明书提取DNA。
1.4 CYP21A2基因全长测序针对CYP21A2基因点突变采用直接测序法检测,参照文献[10, 11],根据CYP21A2和CYP_21A1P基因序列的差别,设计合成了CYP21A2、CYP21A1P基因及嵌合基因的上下游特异性引物,对扩增的3.4 kb DNA进行完全测序,与CYP21A2标准参照序列GenBank M12792.1比对,检测全长CYP21A2基因的点突变。
1.5 多重连接探针扩增技术分析CYP21A2基因缺失或重复突变操作步骤参照荷兰MRC-Holland 公司的SALSA MLPA KIT P050-B2 CAH(Lot 0510)说明书进行,探针和靶序列DNA杂交后通过连接、PCR扩增,产物经毛细管电泳分离得到。收集的数据经Genemapper 4.0(Applied Biosystems)软件分析后得出包括峰高、峰面积、片段长度等一系列数据,根据扩增峰的改变判断靶序列是否有拷贝数的异常或点突变存在,从而分析缺失/重复突变情况。
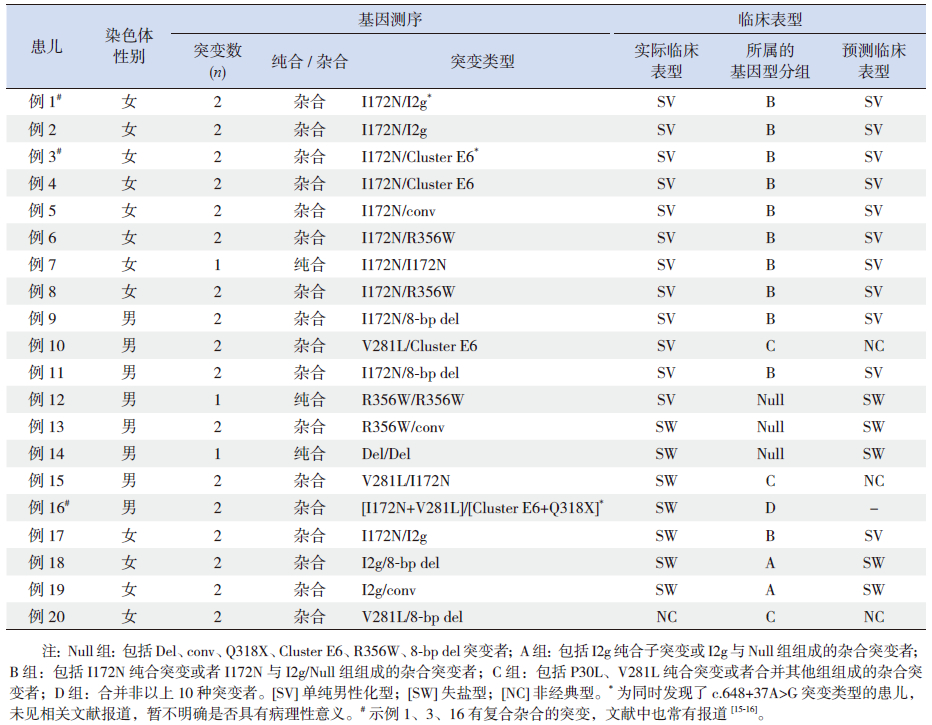
1.6 分组及临床表型预测21-OHD因基因突变的位点不同,可能对酶有不同程度的影响,导致临床表型的差异[12, 13],故按照突变类型将21-OHD患者分成不同的组别,Null组:包括Del、conv、Q318X、Cluster E6、R356W、8-bp del突变者;A组:包括I2g纯合子突变或I2g与Null组组成的杂合突变者;B组:包括I172N纯合突变或者I172N与I2 g/Null组组成的杂合突变者;C组:包括P30L、V281L纯合突变或者合并其他组组成的杂合突变者;D组:合并非以上10种突变者。其中Null组和A组预测为SW型患者;B组预测为SV型患者;C组预测为NC型患者;D组未能进行基因型的预测[14]。
2 结果 2.1 临床症状和体征20例患儿中,1例患儿社会性别为男性,就诊前一直当男童哺养,经腹部B超提示有子宫,做染色体核型分析为46XX方明确性别;1例男性患儿5岁时提前出现第二性征(阴毛早现,阴茎勃起、遗精现象),6岁时即出现变声,生长加速,身高较同龄儿高大(125 cm);所有女性患儿中,均伴有阴蒂肥大,并不同程度逐渐出现皮肤色素沉着、痤疮、胡须旺盛、多毛等男性化表现,其中2例女性患者已行阴蒂切除成形术。
2.2 激素及影像学检查12例SV型患儿血清ACTH及17-OHP水平均明显高于正常值,血清E2水平低于正常值,部分患儿血清T、COR及血钠水平在正常范围内,所有患儿血清LH、FSH及血钾水平均在正常范围(表 1)。
| 表 1 12 例SV 型21-OHD 患儿基础激素水平 |
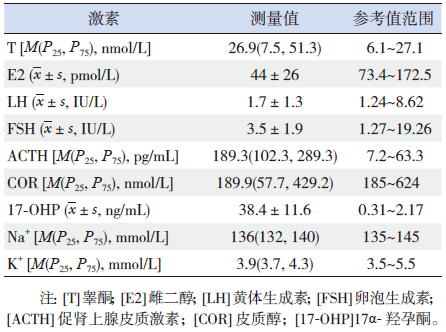
12例SV型患儿中,8例患儿出现骨龄提前;7例患儿有不同程度的肾上腺增生。
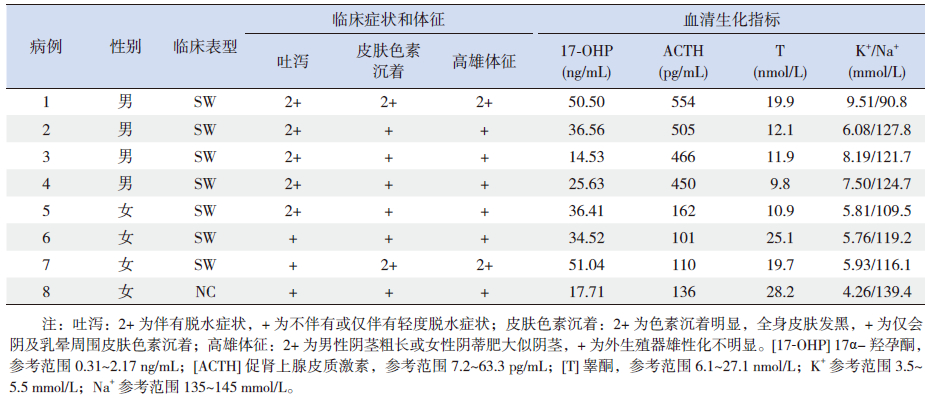
7例SW型患儿有不同程度吐泻、皮肤色素沉着和高雄体征,血清ACTH及17-OHP均高于正常值,且存在明显低钠、高钾。1例NC患儿无吐泻症状,以多毛、初潮延迟就诊,血清ACTH、17-OHP及T水平均高于正常值,怀疑为21-OHD,经同意基因检测后明确诊断。见表 2。
| 表 2 SW 及NC 型21-OHD 患儿临床症状及生化指标 |
20例患儿共发现9种突变,其中8种为已确定的致病突变,分别为Del、conv、I2g、I172N、Cluster E6、8-bp del、V281L、R356W,另1种突变为内含子5上的突变(c.648+37A>G),目前尚未有相关文献报道,暂不明确是否具有病理性意义,该种突变有3例患儿均被检测出。
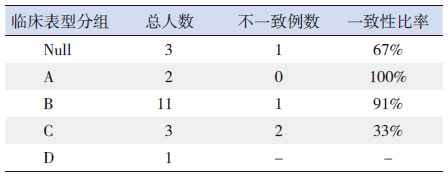
根据基因型的分组进行了临床表型的预测,并与实际的临床表型进行了比对(表 3),并就其一致性进行分析(表 4),结果显示:A、B组中预测的临床表型与实际的临床表型符合率较高,分别为100%和91%,C组的符合率较低,只有33%,D组因未能进行基因型预测而未行一致性分析。
| 表 3 20 例肾上腺皮质增生症患儿基因诊断结果 |
| 表 4 20 例患者基因型和临床表型关联一致性的分析 |
患儿在明确诊断后立即给予醋酸氢化可的松(10~20 mg/m2),早1/2,午、晚各1/4治疗;SW型加用氟氢可的松治疗(每日0.05~0.1 mg,严重失盐者可增至每日0.2 mg);皮质功能危象者先静脉应用氢化可的松(50~100 mg/m2,每日2~3次),同时补充钠盐,待病情稳定后氢化可的松逐渐减量至维持量(每日10~20 mg/m2)。治疗后每3个月复查血电解质、17-OHP、ACTH及T水平;每6~12个月复查骨龄以调整治疗剂量。当患儿出现发热、腹泻等感染情况时,氢化可的松剂量增加2~3倍[17]。坚持长期治疗,使17-OHP及T水平在正常高限或稍高于正常值[18]。替代治疗后,除1例男性SW型患儿因感染时未及时加量甚至有减量,导致肾上腺危象再次就诊,对其纠正肾上腺危象后氢化可的松逐渐减量至维持量继续治疗并定期随访。其余SV型患儿肤色及男性化表现明显好转,SW型患儿酸中毒纠正,血钠、钾恢复正常,生长发育亦基本正常,1例NC型患儿多毛症状改善,并于治疗半年后有月经初潮。1例男性SV型患儿(身高131 cm,骨龄约11岁至11岁半),预测成人终身高为155 cm,明显受损(男孩 <160 cm,女孩<150 cm),向患儿家属建议并经家属知情同意后给予重组人生长激素(rhGH)治疗,有文献显示可改善其生长情况,但尚需更多循证医学依据,不作为常规推荐[19],该患儿应用rhGH治疗1年内,生长速率平均为0.5 cm/月,生长效果欠佳,骨龄评估近12岁(身高137 cm,预测成人终身高仍为155 cm),经与家属协商后未再进行rhGH治疗。对于用药后阴蒂仍肥大未能恢复正常的2例女性SV型患儿,已行阴蒂切除成形术。
3 讨论CAH发病率有明显的种族和地区差别[6],复习文献发现,北京协和医院(9例SW,26例SV,8例NC,均为汉族)的研究中,最常见的突变是I172N(36.0%)[20];中国台湾地区(8例SW,11例SV和1例NC)的研究中,最常见的突变也是I172N(27.5%)[21];与协和医院的研究比较,I172N、I2g和Del的频率无显著差异,R356W频率高于协和医院的研究,所有病人未检测出P30L和V281L突变。新加坡地区的华人21-OHD患者中(SW 12例,SV 8例),最常见的突变也是I172N和I2g,各27.5%,未检测到Exon6 Cluster和V281L[22];与协和医院的研究比较,I172N和I2g频率无显著差别,Del频率显著低于协和医院的研究,而8-bp del和R356W频率显著高于协和医院的研究。
对于NC型21-OHD病人的研究,协和医院[20]与日本Tajima等[23]的研究结果一致,最常见的突变类型是P30L,而白种人中最常见的为V281L,一般在45%~60%[24],犹太人中V218L可高达80%[25]。可见,东西方NC型21-OHD突变特点不同。
本研究中的20例患儿均来自新疆地区,民族均为维吾尔族,其基因突变的频率以I172N(32%,13/40)最常见,同时还发现了内含子5上的一种突变,c.648+37A>G(8%,3/40),而此种基因突变类型目前尚未有相关文献报道,暂不明确是否具有病理性意义。未检出P30L这种基因突变型(只检测出1例Q318X,为复杂杂合突变)。与协和医院[20]、中国台湾[21]、新加坡[22]等不同地区的基因突变频率分布比较可知,新疆地区与台湾地区相同的是均未检测出P30L突变,但新疆地区R356W突变频率(12%,5/40)均较其他地区高,8-bp del的频率(10%,4/40)也较协和医院、台湾地区高,其他突变类型无显著差异。
预测为NC型的3例患儿有1例表现为SV型,1例表现为SW型,可能与不同组合的突变所致酶活性降低程度存在差异相关,另可能存在其他内外源性影响酶活性的因素,例如,新生儿感染可能在非SW型中引起水、电解质紊乱,因此,基因型与临床表型有时也不一致[26]。有报道,基因型对SW型诊断的预测值可达81.8%,对SV型的阳性预测值为94.1%,对NC型的预测值则较低[27]。本研究中基因型对临床表型的阳性预测值高于其他报道[28, 29],考虑可能为本研究纳入的主要为SW型、SV型病例,缺乏NC型病例。由于基因型与表型有较好的相关性,我们可以通过评估患者的表型来验证基因型,反之也可以通过检测患者的基因型来预测疾病的严重程度。本研究中已明确诊断的20例患儿中,其表型均和基因型关联的一致性较高。
CAH是最常见的女性外生殖器性别难辨的病因,也是在未开展新生儿筛查的发展中国家新生儿猝死的常见病因[30]。临床多项研究提示早期诊断、规律治疗且症状控制良好的CAH患儿可有正常的青春期发育[31]。本研究报道的CYP21A2基因突变类型中,复杂杂合突变所占比重较大,且基因突变呈多样性,提示分子诊断需要注意全面性。
综上,经过学者们多年的研究探索,21-OHD型CAH的基因型检测方面已获得了巨大进展,基因型的检出可以预测临床表型的严重程度,同时通过产前诊断可为患者及家庭提供有效的遗传咨询服务。然而,目前尚有许多重要问题有待解决,本研究由于样本量较小,随访过程中有失访的现象,同时可能有自发突变及其他未知突变的存在。但通过对CYP21A2基因突变谱的研究及基因型和表型相关性的分析,可为遗传咨询、基因诊断及产前诊断提供重要的信息。随着研究的深入,将会对中国人21-OHD基因突变有一个更全面的认识。
| [1] | 陆召麟, 卢琳. 先天性肾上腺皮质增生症:非经典型21羟化酶缺陷症的研究进展[J]. 内科急危重症杂志, 2010, 16(1):1-3. |
| [2] | Tsai LP, Lee HH. Analysis of CYP21A1P and the duplicated CYP21A2 genes[J]. Gene, 2012, 506(1):261-262. |
| [3] | Singh RJ. Quantitation of 17-OH-progesterone(OHPG) for diagnosis of congenital adrenal hyperplasia(CAH)[J]. Methods Mol Biol, 2010, 603:271-277. |
| [4] | Ramazani A, Kahrizi K, Razaghiazar M, et al. The frequency of eight common point mutations in CYP21 gene in Iranian patients with congenital adrenal hyperplasia[J]. Iran Biomed J, 2008, 12(1):49-53. |
| [5] | Asanuma A, Ohura T, Ogawa E, et al. Molecular analysis of Japanese patients with steroid 21-hydroxylase deficiency[J]. J Hum Genet, 1999, 44(5):312-317. |
| [6] | Trapp CM, Oberfield SE. Recommendations for treatment of nonclassic congenital adrenal hyperplasia(NCCAH):an update[J]. Steroids, 2012, 77(4):342-346. |
| [7] | 罗飞宏. 先天性肾上腺皮质增生症诊断治疗进展[J]. 中华实用儿科临床杂志, 2015, 30(8):564-569. |
| [8] | Balsamo A, Baldazzi L, Menabò S, et al. Impact of molecular genetics on congenital adrenal hyperplasia management[J]. Sex Dev, 2010, 4(4-5):233-248. |
| [9] | Nimkarn S, New MI. Prenatal diagnosis and treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiency[J]. Mol Cell Endocrinol, 2009, 300(1-2):192-196. |
| [10] | Yu Y, Wang J, Huang X, et al. Molecular characterization of 25 Chinese pedigrees with 21-hydroxylase deficiency[J]. Genet Test Mol Biomarkers, 2011, 15(3):137-142. |
| [11] | Barannik AP, Koltunova AA, Ozolinia LA, et al. A new DNA diagnostic system for the detection of human CYP21 gene mutations associated with adrenal cortex hyperplasia[J]. Bioorg Khim, 2010, 36(3):354-365. |
| [12] | Chen W, Xu Z, Nishitani M, et al. Complement component 4 copy number variation and CYP21A2 genotype associations in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency[J]. Hum Genet, 2012, 131(12):1889-1894. |
| [13] | Baş F, Kayserili H, Darendeliler F, et al. CYP21A2 gene mutations in congenital adrenal hyperplasia:genotypephenotype correlation in Turkish children[J]. J Clin Res Pediatr Endocrinol, 2009, 1(3):116-128. |
| [14] | Krone N, Braun A, Roscher AA, et al. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany[J]. J Clin Endocrinol Metab, 2000, 85(3):1059-1065. |
| [15] | Witchel SF, Smith R, Crivellaro CE, et al. CYP21 mutations in Brazilian patients with 21-hydroxylase deficiency[J]. Hum Genet, 2000, 106(4):414-419. |
| [16] | Wedell A, Thilén A, Ritzén EM, et al. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden:implications for genetic diagnosis and association with disease manifestation[J]. J Clin Endocrinol Metab, 1994, 78(5):1145-1152. |
| [17] | 梁玲, 辛颖. 先天性肾上腺皮质增生症的诊断和治疗进展[J]. 国际儿科学杂志, 2014, 41(1):55-58. |
| [18] | 王昊, 于晓峰. 21-羟化酶缺陷型先天性肾上腺皮质增生症治疗现状[J]. 国际内分泌代谢杂志, 2011, 31(6):393-396. |
| [19] | Longui CA, Kochi C, Calliari LE, et al. Near-final height in patients with congenital adrenal hyperplasia treated with combined therapy using GH and GnRHa[J]. Arq Bras Endocrinol Metabol, 2011, 55(8):661-664. |
| [20] | 张波, 陆召麟, 王玥, 等. 中国人21-羟化酶缺乏症基因型和临床表型特点研究[J]. 遗传学报, 2004, 31(9):950-955. |
| [21] | Ko TM, Kao CH, Ho HN, et al. Congenital adrenal hyperplasia. Molecular characterization[J]. J Reprod Med, 1998, 43(4):379-386. |
| [22] | Loke KY, Lee YS, Lee WW, et al. Molecular analysis of CYP-21 mutations for congenital adrenal hyperplasia in Singapore[J]. Horm Res, 2001, 55(4):179-184. |
| [23] | Tajima T, Fujieda K, Nakae J, et al. Mutations of the CYP21 gene in nonclassical steroid 21-hydroxylase deficiency in Japan[J]. Endocr J, 1998, 45(4):493-497. |
| [24] | Balsamo A, Cacciari E, Baldazzi L, et al. CYP21 analysis and phenotype/genotype relationship in the screened population of the Italian Emilia-Romagna region[J]. Clin Endocrinol(Oxf), 2000, 53(1):117-125. |
| [25] | Weintrob N, Brautbar C, Pertzelan A, et al. Genotypephenotype associations in non-classical steroid 21-hydroxylase deficiency[J]. Eur J Endocrinol, 2000, 143(3):397-403. |
| [26] | Wilson RC, Mercado AB, Cheng KC, et al. Steroid 21-hdroxylase deficiency:genotype may not predict phenotype[J]. J Clin Endocrinol Metab, 1995, 80(8):2322-2329. |
| [27] | Concolino P, Mello E, Patrosso MC, et al. p.H282N and p.Y191H:2 novel CYP21A2 mutations in Italian congenital adrenal hyperplasia patients[J]. Metabolism, 2012, 61(4):519-524. |
| [28] | Marumudi E, Sharma A, Kulshreshtha B, et al. Molecular genetic analysis of CYP21A2 gene in patients with congenital adrenal hyperplasia[J]. Indian J Endocrinol Metab, 2012, 16(3):384-388. |
| [29] | Oriola J, Bosch MZ, Valls C, et al. Association of p.His38Leu, a rare CYP21A2 mutation, with the classical simple virilizing phenotype of 21-hydroxylase deficiency in a 6-year-old boy[J]. Horm Res Paediatr, 2011, 76(3):214-217. |
| [30] | Maiti A, Chatterjee S. Congenital adrenal hyperplasia:an Indian experience[J]. J Paediatr Child Health, 2011, 47(12):883-887. |
| [31] | Hindmarsh PC. Management of the child with congenital adrenal hyperplasia[J]. Best Pract Res Clin Endocrinol Metab, 2009, 23(2):193-208. |