 2016, Vol. 18
2016, Vol. 18



新生儿缺氧性肺动脉高压(hypoxia induced pulmonary hypertension,HPH)是临床常见的急危重症,早期肺血管痉挛,及时治疗可逆;晚期肺血管重塑,临床治疗受限,救治困难,病死率高。本课题组以往的研究表明缺氧诱导因子-1α(HIF-1α)是新生大鼠HPH发病机制中的重要因子[1],近年有研究表明热休克蛋白70(heat shock protein,HSP70)能够在长期缺氧状态下降低HIF-1α的稳定性,促进HIF-1α的降解[2]。然而,目前国内外关于HSP70对HIF-1α降解作用的研究集中在肿瘤等领域[3, 4],而关于HSP70能否在新生儿HPH中延缓HPH的发生发展,未见相关报道。本研究在前期研究的基础上,以HSP70基因为目的基因,借助基因转染技术对HPH新生大鼠肺组织进行外源性基因修饰,以提高肺组织HSP70的表达,观察HSP70基因转染后HPH新生大鼠肺动脉压力、肺血管结构及重塑指标改变的情况,以了解HSP70是否可降低HPH新生大鼠肺动脉压力、减轻其肺血管重塑,为进一步研究新生儿HPH新的治疗方法提供依据。
1 材料与方法 1.1 主要材料与试剂选取7~10日龄健康、清洁Wistar新生大鼠[实验动物许可证号:SCXK(新)2011-0004]128只,体重27±3 g,均由新疆医科大学第一附属医院医学研究中心实验动物研究部提供。氧浓度仪(CY-100B型)由杭州利华科技有限公司提供;小动物呼吸机(HX-200型)由成都泰盟科技有限公司提供;光学显微镜(BX41TF)由日本奥林巴斯公司提供。Ad-HSP70(纯化)及Ad-GFP(纯化)由上海汉恒生物科技有限公司提供,兔抗鼠HSP70多克隆抗体由武汉博士德生物工程有限公司提供,二抗为山羊抗兔IgG抗体-HRP多聚体由中杉金桥生物工程有限公司提供。
1.2 病毒转染及分组将128只Wistar新生大鼠按随机数字表法分为HPH模型组(简称HPH组,n=96)和空白对照组(n=32),其中,HPH组根据转染液不同再分为盐水组、空病毒组(标有绿色荧光信号但未携带目的基因的病毒载体)和病毒+HSP70组(标有绿色荧光信号同时携带目的基因的病毒载体),每组32只大鼠。病毒+HSP70组经大鼠尾静脉注入5 μL 5×1010 PFU/mL AD-HSP70,空病毒组经大鼠尾静脉注入5 μL 5×1010 PFU/mL AD-GFP,盐水组经大鼠尾静脉注入5 μL生理盐水。
1.3 新生大鼠HPH模型的建立按照本课题组前期建立新生大鼠HPH模型的方法[5],将HPH组新生大鼠转染后连同母鼠置于常压低氧舱内,以1.5 L/min的速度将8%氧浓度的氮氧混合气输入低氧舱,用氧浓度测定仪监测舱内氧浓度,使其维持在10%±0.5%,舱内置小风扇,以保证舱内氧浓度均匀。低氧舱内放置钠石灰和无水氯化钙以吸收二氧化碳与水蒸气。温度传感器检测舱内温度使其恒定在 22~24℃。 昼/夜比为12 h/12 h,每天缺氧8 h。空白对照组常规饲养。分别于缺氧3、7、10、14 d检测各组新生大鼠的肺动脉压力及肺血管重塑指标,每个时间点每组处死8只新生大鼠。
1.4 腺病毒定位在HPH新生大鼠肺组织中的判断所有入组新生大鼠测定完肺动脉压力后立即处死取其右中叶肺组织(3~5 mm×3~5 mm× 10~15 mm)。为避免破坏肺组织结构,采集肺组织标本时应注意动作轻巧,避免挤压、损伤肺组织;将切取的右中叶肺组织标本投入生理盐水中洗净血液,然后投入预先配制好的4%多聚甲醛液中固定2 h,再用0.1 mmol/L的PBS液体洗3次,每次5 min;然后将标本置于15%的蔗糖中过夜;最后至于固定液中放入-20℃冰箱备用。将预先备好的标本做冰冻切片,注意避光,并在20 min内放于免疫荧光显微镜下观察,并采集图片。
1.5 免疫组化检测肺组织HSP70取大鼠右下叶肺组织,4%甲醛溶液固定24 h,梯度乙醇脱水,石蜡包埋后切片常规脱蜡,抗原热修复。每只大鼠肺组织制作8张切片。采用兔抗鼠HSP70多克隆抗体及辣根过氧化物酶标记山羊抗兔IgG,免疫组化法测定肺组织HSP70蛋白表达。每个步骤均严格按照试剂盒说明书操作。3,3'-二氨基联苯二胺盐酸盐(0.01 mol/L)显色,磷酸盐缓冲液代替一抗作为阴性对照。阳性标记强度计分:依照细胞阳性着色程度,无着色为0分,淡黄色为1分,棕黄色为2分,棕褐色为3分;每张切片随机观察5个高倍镜视野,依照阳性细胞数量阳性细胞总数为1%~25%计1分,阳性细胞总数为26%~50%计2分,阳性细胞总数为 51%~75%计3分,阳性细胞总数>75%计4分,着色强度计分与阳性面积率计分之积为最后总分:0分(-),1~2分(+),3~4分(2+),>4分(3+)。
1.6 平均肺动脉压力的测定各组新生大鼠经氯胺酮(75 mg/kg)、阿托品(0.375 mg/kg)和地西泮(7.5 mg/kg)混合液腹腔内注射麻醉后,固定、备皮、消毒气管及胸部皮肤,行气管插管,给予机械通气,小动物呼吸机参数设置为呼吸频率100~110次/min、吸呼比1 : 1.5、潮气量5~7 mL/min。监测实验新生大鼠尾部血氧饱和度,使其维持在85%~95%。从胸骨右缘行U型切口打开胸腔(右至胸骨右缘约0.3 cm,左至胸骨左缘约0.5 cm,下至剑突下缘),充分暴露肺动脉根部,用4.5号针头逆血流方向小心刺入肺动脉根部,针头的另一端连接记录仪压力传感器,记录平均肺动脉压(mean pulmonary arterial pressure,mPAP)。
1.7 肺血管重塑指标的检测取Wistar 新生大鼠右上叶肺组织,4%多聚甲醛固定1周。常规石蜡包埋,连续切片,制备好的石蜡切片常规脱蜡脱水,每只大鼠随机选3张肺组织切片行苏木精-伊红(HE)染色,每张切片随机选取与呼吸性细支气管及肺泡管伴行的横断面积较圆、直径为5~10 μm的肺小动脉3支,在光镜下观察形态学变化。用病理图像分析软件测定肺小动脉中层壁厚(MT)、肺小动脉外径(ED)、管壁中层横截面积(MA)和血管总横截面积(TAA),根据公式MT%=MT/ED×100%、MA%=MA/TAA×100%计算,将MT%、MA%作为肺血管重塑指标。
1.8 统计学分析采用SPSS 20.0统计软件对数据进行统计学分析。符合正态分布的计量资料采用均数±标准差(x±s)表示,多组间比较采用One-Way ANOVA分析,组间两两比较采用SNK-q检验;不满足正态分布的计量资料采用中位数(四分位间距)[P50(P25,P75)]表示,多组间比较采用 Kruskal-Wallis H检验,组间两两比较采用Nemenyi检验;等级资料采用例数表示,多组间比较采用秩转换后的Kruskal-Wallis H检验,组间两两比较采用Nemenyi检验。P<0.05为差异有统计学意义。
2 结果 2.1 腺病毒在HPH新生大鼠肺组织中的定位取空病毒组及空白对照组肺组织冰冻切片于免疫荧光显微镜下观察,发现缺氧3、7、10 d时空病毒组肺组织均可见免疫荧光;缺氧14 d时未见明显免疫荧光(图 1)。表明经尾静脉途径注射的腺病毒成功定位于新生大鼠肺组织,随着缺氧时间延长,腺病毒在肺组织内逐渐衰减。
 |
图 1 免疫荧光法观察空病毒组肺组织中腺病毒定位(GFP,×200) 空病毒组肺组织在缺氧3 d 时可见明显 绿色荧光,随着时间延长,空病毒组绿色荧光逐渐减退,至缺氧14 d 时绿色荧光消失。 |
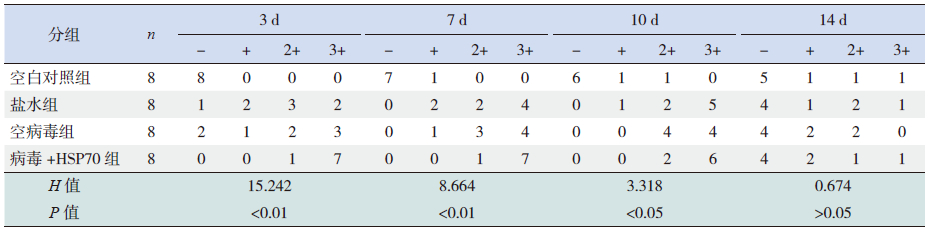
缺氧3、7、10 d 盐水组、空病毒组、病毒+ HSP70组肺组织HSP70表达均强于空白对照组(P<0.01),其中病毒+HSP70组HSP70表达强于与盐水组和空病毒组(P<0.01),但在盐水组和空病毒组间比较差异无统计学意义(P>0.05);缺氧14 d时各组间HSP70表达强度比较差异无统计学意义(P>0.05)。见图 2,表 1。
 |
图 2 新生大鼠肺组织中HSP70 表达(DAB 显色,×200) 缺氧3、7、10 d 病毒+HSP70 组HSP70 表达强于其余3 组, 随着缺氧时间延长,HSP70 逐渐衰减,至缺氧14 d 时,4 组间HSP70 表达无明显差异。HSP70 阳性表达呈棕黄色。 |
| 表 1 4 组新生大鼠HSP70 免疫组化反应强度比较 (鼠数) |
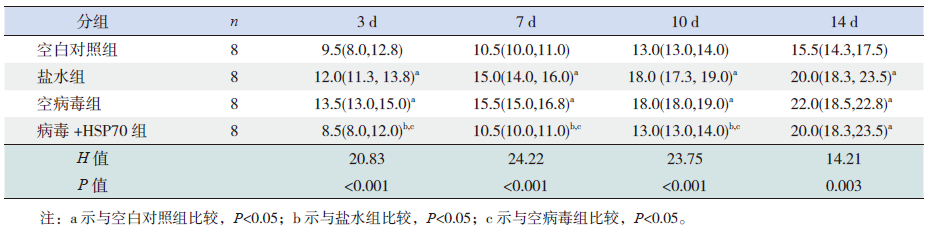
盐水组及空病毒组缺氧3、7、10、14 d时的mPAP水平比同日龄空白对照组均显著增高(P<0.05)。病毒+HSP70组缺氧3、7、10 d时的mPAP水平与同日龄空白对照组比较差异无统计学意义(P>0.05);与同日龄盐水组及空病毒组比较显著降低(P<0.05)。病毒+HSP70组缺氧14 d时mPAP水平与同日龄盐水组及空病毒组比较差异无统计学意义(P>0.05),但高于空白对照组(P<0.05)。见表 2。
| 表 2 4 组新生大鼠mPAP 比较 [P50(P25,P75)] |
空白对照组新生大鼠肺小动脉管壁薄;盐水组和空病毒组新生大鼠缺氧3 d时肺小动脉管壁均明显增厚,缺氧7 d时,呼吸性细支气管水平肺小动脉平滑肌层增厚,管壁增厚,官腔缩小,缺氧10 d及14 d时可见肺小动脉中层平滑肌增生,管壁增厚,官腔更加狭小;病毒+HSP70组在缺氧3、7、10 d时肺小动脉中层平滑肌未见明显增生,管壁增厚不明显,官腔狭小不显著,在缺氧14 d时肺小动脉中层平滑肌增生,管壁增厚,官腔狭小,提示病毒+HSP70组肺血管结构改变较盐水组和空病毒组延迟并且程度较轻。见图 3。
 |
图 3 缺氧后新生大鼠肺组织形态学(苏木素- 伊红染色,×200) 空白对照组新生大鼠肺小动脉管壁薄。盐水 组和空病毒组随着缺氧时间延长,肺小动脉管腔逐渐缩小,管壁增厚。病毒+HSP70 组在缺氧3、7、10 d 时肺小动脉管腔无 明显缩小,管壁增厚不显著;在缺氧14 d 时肺小动脉管壁增厚,官腔狭小。 |
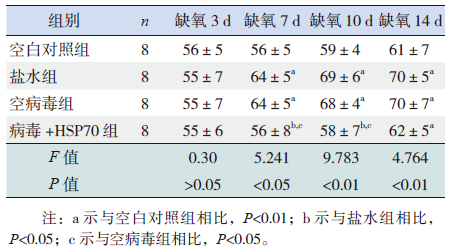
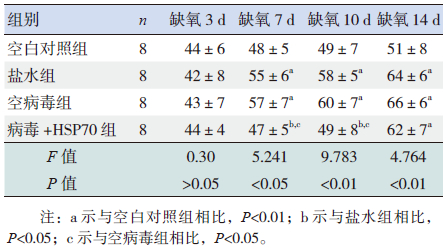
盐水组和空病毒组在缺氧3 d时MT%和MA%与空白对照组比较差异无统计学意义(P>0.05);缺氧7、10、14 d时MT%和MA%较空白对照组明显增高(P<0.05);但此两组间比较差异无统计学意义(P>0.05),提示此盐水组和空病毒组缺氧7 d时开始出现肺血管重塑。病毒+HSP70组在缺氧3、7、10 d时MT%和MA%与空白对照组比较差异无统计学意义(P>0.05);在缺氧7、10 d时MT%和MA%低于盐水组和空病毒组(P<0.05);缺氧14 d时与盐水组和空病毒组比较差异无统计学意义(P>0.05),但高于空白对照组(P<0.05)。提示病毒+HSP70组肺血管重塑在缺氧14 d时出现,较盐水组和空病毒组晚。见表 3~4。
| 表 3 4 组新生大鼠MA% 变化 ( x±s,%) |
| 表 4 4 组新生大鼠MT% 变化 ( x±s,%) |
新生儿HPH,救治困难,病死率高,其发病机制有待进一步研究。本课题组前期研究发现,HIF-1α在新生儿HPH的发病机制中发挥了关键作用[5],通过促进下游靶基因的表达[6],使肺血管内皮损伤,肺血管痉挛及重塑,参与肺动脉高压的形成[7]。HSP又称为应激蛋白或热应激蛋白,具有高度保守性,它普遍存在于整个生物界,几乎所有的细胞均能合成HSP。HSP70在正常情况下表达量很少,而在缺氧等应激刺激状态下表达明显升高,并且在细胞浆和细胞核内均有表达[8, 9]。主要参与细胞内蛋白质的合成、运输、折叠和蛋白质的降解调控过程,可提高细胞对应激原的耐受性,使细胞维持正常的生理功能[10]。近年又有研究表明HSP70能够在长期缺氧状态下降低HIF-1α的稳定性,促进HIF-1α的降解[2],从而降低肺动脉压力,减轻肺血管重塑。相关研究局限于成人肿瘤等领域,在新生儿HPH是否有同样作用国内外未见相关报道。
本研究中,盐水组及空病毒组缺氧新生大鼠在缺氧3、7、10、14 d时mPAP均高于空白对照组,提示肺动脉高压形成,造模成功。另外,经大鼠尾静脉途径注射Ad-HSP70对HPH新生大鼠肺组织进行外源性基因修饰,病毒+HSP70组HSP70表达较其余3组明显增多,表明可以利用重组腺病毒介导外源性HSP70基因转染至HPH新生大鼠肺组织中,转染成功,提高了肺组织中HSP70的表达,为本课题的研究提供了保证。
HPH的形成主要与低氧性肺血管收缩和肺血管重塑有关。目前认为肺血管重塑是肺动脉高压持续发展的关键因素,决定着肺动脉高压的持续存在与发展。本课题组前期研究发现,Wistar大鼠乳鼠在缺氧3~5 d时为肺血管痉挛阶段,此过程为可逆性改变,若缺氧及时纠正,可以避免肺血管重塑;而缺氧7 d以上,肺血管重塑难以避免,将发生不可逆改变,预后不良[11]。本研究结果显示,盐水组及空病毒组在缺氧3 d时肺动脉处于收缩状态,出现mPAP增强;在缺氧7 d时肺动脉结构发生改变,主要包括肌型小动脉中膜增生肥厚、无肌型动脉肌化等,导致肺血管重塑的改变[12],mPAP进一步增强。
本研究结果显示,盐水组及空病毒组在缺氧3、7、10 d时HSP70阳性率高于空白对照组,考虑与缺氧后HSP70转录因子明显激活一致,此为内源性表达结果,而在缺氧14 d,三组间HSP70阳性率表达差异无统计学意义,表明随肺组织缺氧时间延长,细胞结构和功能破坏,10 d内肺组织的应激和抗损伤能力较强,因而可产生HSP70以进行抗损伤和修复,随着缺氧时间延长,肺组织细胞受损加重,能够产生HSP70的能力下降。
本研究显示,在缺氧3、7、10 d时病毒+ HSP70组HSP70在肺组织中充分表达,此组mPAP与盐水组、空病毒组比较明显降低,肺血管未见明显重塑;缺氧14 d时,HSP70在新生大鼠肺组织内代谢完全,此时其mPAP较空白对照组明显增高,肺血管出现重塑;说明腺病毒介导的HSP70能减轻肺血管重塑,降低肺动脉压力。Gogate等[8]发现HSP70在低氧条件下能够促进HIF-1α通过蛋白酶体途径降解,而利用HSP抑制剂KNK437抑制HSP70的功能后,HIF-1α的降解减少、活性增加。还有研究发现HSP70基因结构中含有缺氧诱导因子结合位点,很有可能是HIF-1α的下游靶基因,缺氧激活HIF-1α的转录活性,同时促进HSP70表达增加,表达增加的HSP70反过来激活HIF-1α的泛素化降解,成为机体细胞对缺氧反应的负反馈通路[4],进一步说明在缺氧状况下HSP70对HIF-1α具有泛素化降解作用,从而降低肺动脉压力,具体作用机制还需进一步研究。
本研究发现HSP70可能可以降低肺动脉压力,减轻肺血管重塑,这可能与HSP70促进HIF-1α的降解有关,本课题组正在做进一步研究。
| [1] | 周英, 王冬梅, 朱艳萍, 等. 缺氧诱导因子-1α在新生大鼠缺氧性肺动脉高压中的作用及其与肺血管重塑的关系[J]. 中华围产医学杂志, 2014, 17(4):260-266. |
| [2] | Luo W, Zhong J, Chong R, et al. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor(HIF)-1alpha but not HIF-2alpha[J]. J Biol Chem, 2010, 285(6):3651-3663. |
| [3] | van de Sluis B, Groot AJ, Vermeulen J, et al. COMMD1 promotes pVHL and O2-independent proteolysis of HIF-1alpha via HSP90/70[J]. PLoS One, 2009, 4(10):e7332. |
| [4] | 夏丽敏, 田德安, 张琼, 等. 缺氧诱导热休克蛋白70-2表达的分子机制[J]. 中华肝病杂志, 2009, 17(3):207-212. |
| [5] | 桑葵, 周英, 李明霞. 缺氧诱导因子1α及其调控因子在新生大鼠缺氧性肺动脉高压发病机制中作用的研究[J]. 中华儿科杂志, 2012, 50(12):919-924. |
| [6] | Wang L, Zhou Y, Li M, et al. Expression of hypoxia-inducible factor-1α, endothelin-1 and adrenomedulin in newborn rats with hypoxia-induced pulmonary hypertension[J]. Exp Ther Med, 2014, 8(1):335-339. |
| [7] | 贺洪军, 戴爱国. 肺心汤对低氧性肺动脉高压模型大鼠低氧诱导因子-1α及血管内皮生长因子的影响[J]. 中国中西医结合杂志, 2012, 32(5):676-680. |
| [8] | Gogate SS, Fujita N, Skubutyte R, et al. Tonicity enhancer binding protein(TonEBP) and hypoxia-inducible factor(HIF) coordinate heat shock protein 70(Hsp70) expression in hypoxic nucleus pulposus cells:role of Hsp70 in HIF-1α degradation[J]. J Bone Miner Res, 2012, 27(5):1106-1117. |
| [9] | 王燕婷, 梁建辉. 精神活性物质对热休克蛋白70表达的影响[J]. 生理科学进展, 2012, 43(1):66-70. |
| [10] | Tian YM, Yeoh KK, Lee MK, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors[J]. J Biol Chem, 2011, 286(15):13041-13051. |
| [11] | 桑葵, 周英, 李明霞. 缺氧性肺动脉高压新生大鼠肺血管重塑的研究[J]. 中国当代儿科杂志, 2012, 14(3):210-214. |
| [12] | 杜军保, 唐朝枢. 肺动脉高压[M]. 北京:北京大学医学出版社, 2010:24-90. |