 2016, Vol. 18
2016, Vol. 18



2. 解放军第302 医院中西医结合肝病中心, 北京 100039
先天性肝纤维化(congenital hepatic fibrosis,CHF)是一种胆管上皮细胞病变引起的疾病,其特征是胆管出现类似胆管板畸形改变和进行性胆管周围纤维化[1]。CHF主要在儿童和青少年中起病[2, 3]。根据其临床表现,CHF可分为门脉高压型CHF、胆管炎型CHF、门脉高压和胆管炎混合型CHF和隐匿型CHF[4]。CHF是较少见病,病例数较少,文献中对儿童CHF发病特点报道较少,局限于个案报道,本文拟通过对60例确诊CHF患儿(小于18岁)的回顾性研究,探讨儿童CHF临床分型的特点。
1 资料与方法 1.1 一般资料2002年1月至2015年6月期间在解放军第302医院住院并经肝穿刺组织学病理诊断确诊为CHF的患儿60例,其中男40例,女20例,年龄均小于18岁。26例为门脉高压型,3例为胆管炎型,30例为混合型,1例为隐匿型。因胆管炎型和隐匿型患儿过少,难以进行统计分析,故予以剔除。
1.2 研究指标采集26例门脉高压型和30例混合型患儿的性别、年龄、临床表现、体征、实验室检查、影像特点等临床资料进行回顾性研究。其中实验室检查指标包括血清白蛋白(ALB)、丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(AST)、总胆红素(TBIL)、碱性磷酸酶(ALP)、ν谷氨酰转肽酶(GGT)、亮氨酸氨基肽酶(LAP)、总胆汁酸(TBA)等肝功能指标及白细胞总数(WBC)、单核细胞绝对值(M)、红细胞总数(RBC)、血红蛋白(HGB)、血小板总数(PLT)、血小板平均体积(MPV)、凝血酶原活动度(PTA)和国际标准化比值(INR)。
1.3 组织学B超引导下采用快速穿刺法取肝组织。标本长1.3~2.0 cm,镜下包括至少6个汇管区。常规石蜡包埋,行苏木精-伊红染色及Masson染色。由解放军第302医院病理科采用统一标准一次性阅片,病理诊断参照文献标准[2]。
1.4 分型CHF分型依据Desmet[5]提出的分为4个临床型:(1)门脉高压型:患者有肝脾肿大、腹水、消化道出血、食道胃底静脉曲张或脾功能亢进(WBC计数、PLT计数减少)等门脉高压症表现。(2)胆管炎型:患者有胆汁淤积表现,无门脉高压症状,病理提示存在胆管炎。(3)混合型:患者同时有门脉高压症和胆管炎表现。(4)隐匿型:患者经肝穿刺病理证实存在CHF,无门脉高压症和胆管炎表现。
1.5 统计学分析应用SPSS 19.0统计软件对数据进行统计学分析,非正态分布的计量资料以中位数(四分位间距)[M(P25,P75)]表示,两组间比较采用Wilcoxon秩和检验。计数资料以百分率(%)表示,两组间比较采用卡方检验。P<0.05为差异有统计学意义。
2 结果 2.1 年龄及性别门脉高压型患儿中,男20例,女6例,年龄为11.00(6.75,14.50)岁;混合型患儿中,男16例,女14例,年龄为11.50(6.75,16.00)岁;两组在年龄和性别之间比较差异无统计学意义(分别χ2=11.717、3.376,P>0.05)。
2.2 症状体征在56例患儿中,上消化道出血(呕血或黑便)最为常见,达25例(45%),以下依次为乏力10例(18%)、发热9例(16%)、腹胀8例(14%)、腹痛7例(12%)、黄疸6例(11%)、食欲减退5例(9%),无症状者11例(20%)。上述症状中仅发热和黄疸在混合型患儿中的发生率高于门脉高压型患儿(P<0.05),其余症状发生率在两型患儿中比较差异无统计学意义(P>0.05),见表 1。
| 表 1 两组临床表现的比较 [ 例(%)] |
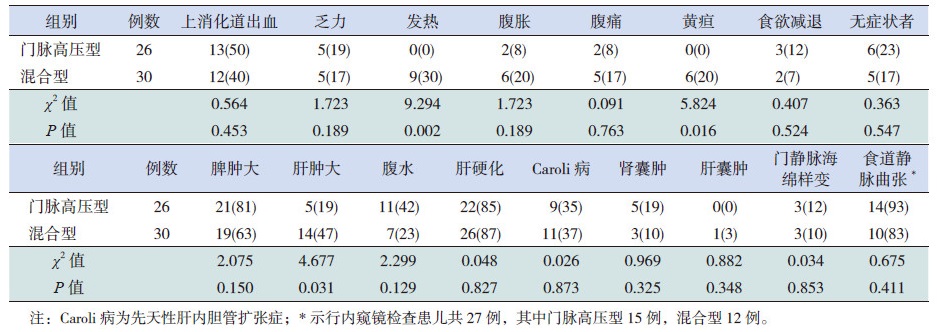
体查中发生脾肿大患儿最多,达40例(71%),肝肿大19例(34%)次之,腹水18例(32%),其中混合型患儿肝肿大发生率高于门脉高压型患儿(P<0.05),见表 1。
2.3 影像学及内镜检查影像学检查中肝硬化是最为常见的,48例(86%)患者提示肝硬化,20例(36%)患者影像学提示合并先天性肝内胆管扩张症,合并肾囊肿和肝囊肿的患者分别为8例(14%)和1例(2%),6例(11%)患者出现门静脉海绵样变。上述影像学结果在两组间比较差异均无统计学意义(P>0.05),见表 1。
共有27例患者进行内窥镜检查,24例患者发现食道静脉曲张,但在两组之间比较差异无统计学意义(P>0.05),见表 1。
2.4 家族史仅1例(2%)患儿有先天性肝纤维化家族史。
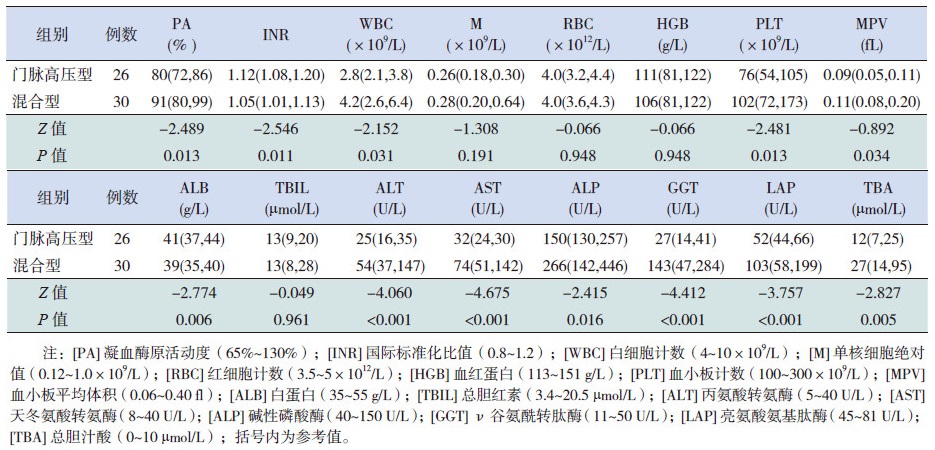
2.5 实验室检查凝血功能中,门脉高压型和混合型中PA和INR均在正常范围内,但在两组之间比较差异有统计学意义(P<0.05)。血常规中,WBC和PLT在门脉高压型患儿中出现下降,在混合型患儿中处于正常范围;M、RBC和MPV在门脉高压型和混合型患儿中均正常;HGB在门脉高压型和混合型患儿中均出现下降;其中WBC、PLT和MPV在两组间比较差异有统计学意义(P<0.05)。生化检查中,ALB和TBIL在两型患儿中均处于正常水平;在门脉高压型患儿中,除了TBA出现轻度升高外,ALT、AST、ALP、GGT和LAP均处于正常范围;而在混合型患儿中ALT、AST、ALP、GGT、LAP和TBA均出现了升高;其中ALB、ALT、AST、ALP、GGT、LAP和TBA等指标在两组间比较差异均有统计学意义(P<0.05)。见表 2。
| 表 2 两组凝血功能、血常规和生化检查结果比较 [M(P25,P75)] |
CHF的发病年龄差异很大,从童年早期至50岁或60岁均可发病[6, 7],但儿童期发病为主。在本研究中,门脉高压型中位年龄在11岁,混合型中位年龄在11.5岁,符合这一发病规律,并提示在此年龄段以门脉高压为表现的患儿,要注意除外CHF。CHF的特征性表现是出现发育不良的肝内胆管,这是由于发育期间小叶间胆管受干扰而出现胆管板畸形导致的[2]。排列于胆管系统的上皮细胞——胆管上皮细胞是CHF的主要靶点[8]。生理条件下,胆管上皮细胞通过基底膜和激素调节活动促进了最终胆汁成分和量的分泌[9]。胆管上皮细胞受损,使胆汁生成的定性/定量改变,出现胆汁淤积,这被称为胆管细胞性胆汁淤积。胆汁酸在肝内合成及分泌,其血清升高是胆汁淤积敏感和早期肝特异性指标[10]。因此,在门脉高压型和混合型中TBA高于正常范围,表明CHF这两种类型均属于胆汁淤积性肝病。
在胆汁淤积时,ALT和AST一般不升高[10],ALT或AST等转氨酶升高表明存在肝细胞损伤[11]。本研究中混合型ALT和AST均出现升高,表明肝细胞损伤,而门脉高压型ALT和AST正常,表明无肝细胞损伤。早期研究认为在胆汁淤积性肝病中,胆汁酸直接毒性诱导的细胞凋亡是肝细胞损伤的主要原因[12]。但近期研究证实,活体内胆汁酸不是直接导致胆汁淤积性肝损伤的原因,胆汁酸是作为炎症刺激剂,直接活化肝细胞中信号转导途径来生成促炎性介质,募集了白细胞等炎性细胞介导胆汁淤积期间肝损伤[13]。因此,虽然TBA在两种类型患儿中均出现升高,但由于门脉高压型患儿中TBA是轻度升高,混合型患儿中TBA是中度升高,两者之间存在统计学差异,对白细胞等炎性细胞的募集作用也产生了差异,致使混合型患儿出现了比门脉高压型患儿更明显的肝细胞损伤。胆汁淤积等应激诱导细胞损伤引起的是无菌性炎症[14],因此,在本研究中,门脉高压型和混合型患儿中WBC均未高于正常范围上限,这与CHF基本病理变化,即胆管板畸型常伴非化脓性破坏性胆管炎[2]是相一致的。
在胆汁淤积性肝损伤动物模型中发现,白细胞积聚介导胆汁淤积性肝细胞损伤是血小板依赖性的[15]。研究表明血小板除止血功能之外,还参与肝脏的多个病理过程,包括炎症和再生。清除和抑制血小板,可明显减轻白细胞介导的肝细胞损伤[16]。混合型中由于存在足够量的血小板,促使了白细胞积聚于肝脏,导致ALT、AST出现升高,而在门脉高压型中由于血小板数不足,白细胞在肝内积聚减少,肝功能未受明显损伤。虽然混合型和门脉高压型相比,肝功能损伤更为明显,但混合型肝功能损伤是轻度的,因此肝脏合成功能虽受损,白蛋白与门脉高压型相比有下降,但白蛋白仍处于正常范围。在临床症状方面,由于混合型存在胆管炎,发热和黄疸症状也较门脉高压型更多见。血小板依赖性白细胞积聚作用可由MPV进一步证实。MPV是血小板活化的标志[17]。血小板与白细胞交互作用,可进一步活化血小板。混合型和门脉高压型中MPV虽处于正常范围,但混合型中MPV明显高于门脉高压型,表明混合型中血小板活化要比门脉高压型明显。活化血小板与白细胞的交互作用,可扩增生理和病理状态下的凝血级联反应[18]。因此,在混合型中,PA高于门脉高压型,INR低于门脉高压型,说明混合型凝血级联反应得以进一步扩增。
在肝病中,血清ALP升高是胆汁淤积的标志[19]。胆汁酸是胆道ALP分泌的主要决定因素[19],混合型中因TBA明显高于门脉高压型,因此在混合型患儿中ALP出现明显升高。在原发性胆汁性肝硬化患者中发现,血清ALP水平出现明显升高时,常不能对熊去氧胆酸治疗应答,而血清ALP低水平患者经熊去氧胆酸治疗,具有极好的长期预后[19]。在CHF患者中,血清ALP是否也是病情严重程度的标志尚需要进行前瞻性研究。在儿童中,血清ALP活性相当高,这与骨生长相关[19]。除了ALP外,胆汁淤积的酶标记物还包括了GGT、LAP和5'-核苷酸酶[20]。在临床实践中,区分升高的来源是通过检测其他酶来完成。由于在骨病中未发现LAP升高,因此在胆汁淤积中,LAP要比ALP和GGT更具特异性[20]。但在胆汁淤积中,GGT要比LAP敏感9倍,比ALP敏感6倍[20],因此,在CHF中,ALP、LAP和GGT常联合使用来区分ALP升高是否源于肝胆病变。
遗传学进展证实CHF是一个由PKHD1基因突变导致的遗传性相关胆管病,PKHD1基因可编码fibrocystin/polyductin(FPC),fibrocystin位于啮齿动物胆管上皮细胞的纤毛,参与了肾集合小管和胆管分化[21]。在肝脏中,FPC缺乏会导致胆管微小错构瘤形成以及胆管节段性扩张,伴进行性门脉纤维化,临床上导致相关的门脉高压[1]。最近研究证实FPC缺陷的胆管上皮细胞经募集巨噬细胞可促进CHF中门脉性纤维化的形成,经氯膦酸清除巨噬细胞会导致门脉性纤维化和门脉高压减轻[1]。在本研究中,由于门脉高压型和混合型均存在着FPC缺陷的胆管上皮细胞,且参与门脉性纤维化形成的单核细胞(巨噬细胞属于单核细胞)在两型中无差异,因此在两型中最为明显的改变是80%以上患者影像学均提示肝硬化,且在脾肿大、上消化道出血、食道静脉曲张、腹水等门脉高压表现以及合并Caroli病、肾囊肿和肝囊肿等方面无统计学差异。因出现上消化道出血,两组中HGB均出现下降。肝脏大小与剩余肝实质团或肝脏再生能力相关,混合型中肝肿大较门脉高压型明显,说明混合型中剩余肝实质团比门脉高压型多,或其再生能力要比门脉高压型更强。
血管和胆管的发育是密切相关的。门静脉可引导胆管板发育。胆管板似乎可诱导肝动脉形成肝内分支。肝动脉分支似乎可引起周围型胆管渗入门静脉周围的间充质中。由于存在这种密切关系,血管和肝内胆管的异常经常相重叠[6]。因此,在本研究中,在门脉高压型和混合型患儿中均见到门脉海绵样变性,但在两型患儿中差异无统计学意义。
总之,本研究通过对儿童CHF临床分型的研究表明,门脉高压型和混合型较常见,胆管炎型和隐匿型罕见,门脉高压型和混合型中以肝硬化、肝脾肿大等门脉高压表现明显,但混合型肝损伤更为明显。
| [1] | Locatelli L, Cadamuro M, Spirli C, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis[J]. Hepatology, 2016, 63(3): 965-982. |
| [2] | Desmet VJ. What is congenital hepatic fibrosis?[J]. Histopathology, 1992, 20(6): 465-477. |
| [3] | 吴欣, 李忠斌, 刘红虹, 等. 35例先天性肝纤维化患者的临床及病理特点[J]. 胃肠病学和肝病学杂志, 2013, 22(6): 529-532. |
| [4] | Arnon R, Rosenberg HK, Suchy FJ. Caroli disease, caroli syndrome, and congenital hepatic fibrosis[M]. Farmington: Humana Press, 2010: 331-358. |
| [5] | Desmet VJ. Ludwig symposium on biliary disorders—part I. Pathogenesis of ductal plate abnormalities[J]. Mayo Clin Proc, 1998, 73(1): 80-89. |
| [6] | Veigel MC, Prescott-Focht J, Rodriguez MG, et al. Fibropolycystic liver disease in children[J]. Pediatr Radiol, 2009, 39(4): 317-327. |
| [7] | 吴欣, 周超, 罗生强. 先天性肝纤维化不同分型的临床特征—75例分析[J]. 肝脏, 2014, 19(7): 479-482, 490. |
| [8] | Park SM . The crucial role of cholangiocytes in cholangiopathies[J]. Gut Liver, 2012, 6(3): 295-304. |
| [9] | Masyuk AI, Masyuk TV, LaRusso NF. Cholangiocyte primary cilia in liver health and disease[J]. Dev Dyn, 2008, 237(8): 2007-2012. |
| [10] | 胆汁淤积性肝病诊断治疗专家共识 2015年更新专家委员会. 胆汁淤积性肝病诊断治疗专家共识: 2015年更新[J]. 临床肝胆病杂志, 2015, 13(10): 1563-1574. |
| [11] | Karpen SJ. Update on the etiologies and management of neonatal cholestasis[J]. Clin Perinatol, 2002, 29(1): 159-180. |
| [12] | Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury[J]. World J Gastroenterol, 2012, 18(36): 4985-4993. |
| [13] | Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis[J]. Am J Pathol, 2011, 178(1): 175-186. |
| [14] | Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts[J]. J Gastroenterol Hepatol, 2011, 26 Suppl 1: 173-179. |
| [15] | Laschke MW, Dold S, Menger MD, et al. Platelet-dependent accumulation of leukocytes in sinusoids mediates hepatocellular damage in bile duct ligation-induced cholestasis[J]. Br J Pharmacol, 2008, 153(1): 148-156. |
| [16] | Sullivan BP, Wang R, Tawfik O, et al. Protective and damaging effects of platelets in acute cholestatic liver injury revealed by depletion and inhibition strategies[J]. Toxicol Sci, 2010, 115(1): 286-294. |
| [17] | Park Y, Schoene N, Harris W. Mean platelet volume as an indicator of platelet activation: methodological issues[J]. Platelets, 2002, 13(5-6): 301-306. |
| [18] | Vieira-de-Abreu A, Campbell RA, Weyrich AS, et al. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum[J]. Semin Immunopathol, 2012, 34(1): 5-30. |
| [19] | Poupon R. Liver alkaline phosphatase: a missing link between choleresis and biliary inflammation[J]. Hepatology, 2015, 61(6): 2080-2090. |
| [20] | Kuntz E, Kuntz HD. Cholestasis[M]//Kuntz E, Kuntz HD. Hepatology principles and practice. 2nd. Wetzlar: Springer Berlin Heidelberg, 2006: 227-242. |
| [21] | Lazaridis K N, Strazzabosco M, Larusso N F. The cholangiopathies: disorders of biliary epithelia[J]. Gastroenterology, 2004, 127(5): 1565-1577. |