 2016, Vol. 18
2016, Vol. 18

根据病因的不同,戊二酸尿症分为戊二酸尿症1型、2型和3型,其中戊二酸尿症1型(MIM#231670)是最常见的有机酸代谢性脑病之一,是一种常染色体隐性遗传病,是由于戊二酰辅酶A脱氢酶(glutaryl-CoA dehydrogenase,GCDH)活性降低或缺陷导致的有机酸尿症,生化特点为体内戊二酸、3-羟基戊二酸蓄积,引起纹状体等神经核团损害,导致神经退行性疾病[1, 2, 3]。自1975年报道第一例戊二酸尿症1型以来,至今国内外已报道病例超过400例[4, 5, 6]。该病在全球发病率约为1 : 100 000[1];美国发病率约1 : 50 000[2];瑞典1 : 30 000[3];我国浙江省报道的2008~2010年发病率为1/64 708[7],台湾2000~2009年自1 495 132名新生儿检出13例、发病率为1/115 010[4]。戊二酸尿症1型患者的临床表现多样,可引起急性脑病和进行性神经损害,早期诊断及合理治疗可有效降低神经系统致残率,故提高对本病的临床和实验室特点的认识对改善预后有重要意义。现就戊二酸尿症1型的临床与实验研究进展进行综述。
1 病因、发病机制戊二酰辅酶A脱氢酶位于线粒体基质,在线粒体内将戊二酰辅酶A转化成巴豆酰辅酶A。编码戊二酰辅酶A脱氢酶GCDH基因突变导致酶活性降低或缺陷,赖氨酸、羟赖氨酸和色氨酸代谢障碍,戊二酰辅酶A过度堆积,患者体内戊二酸、3-羟基戊二酸浓度显著升高[1, 2, 3]。此外,戊二酰辅酶A可与游离肉碱发生酯化反应,患者血中戊二酰肉碱升高,游离肉碱消耗增加,引起继发性肉碱缺乏。患者神经系统损害与戊二酸和3-羟基戊二酸的神经毒性有关[1, 2, 3]。
2 临床表现戊二酸尿症1型临床表现具有异质性,胎儿至成年均可发病,轻者无临床症状,重者表现为严重的急性脑病,甚至死亡[5, 6, 8]。大多数患者出生时即表现出头围大或增长过快。婴幼儿时期是体格、大脑快速发育的关键时期,原本表现“正常”的戊二酸尿症1型患儿常因高蛋白饮食、发热、感染、疲劳、疫苗接种等诱因引发急性脑病,出现运动倒退、肌张力低下、癫癎,导致肢体僵硬、肌张力不全和锥体外系等症状[8, 9]。患者认知功能通常正常[10, 11]。部分患者发病较晚,症状相对较轻,可表现为头痛、视力下降、运动障碍等,发病前智力、运动水平均正常[8]。目前已知的发病年龄最晚的戊二酸尿症1型患者为陈靖等报道的一例我国男性患者,于51岁时因暴饮暴食、酗酒诱发急性脑病[12]。
额叶、颞叶发育不全、双侧外侧裂增宽、双侧基底节对称性改变和脑白质低密度灶是戊二酸尿症1型患者常见的神经影像学表现[8, 13]。部分患者早期无明显的神经影像学改变,随着病情进展可出现基底节损害或脑白质异常,颞部脑萎缩、双侧裂池明显扩大提示病情加重[13, 14, 15]。磁共振波谱分析对研究戊二酸尿症1型患者的脑神经代谢功能具有参考价值[16]。基底节区乳酸堆积提示患者脑能量代谢受损,N-乙酰天门冬氨酸降低提示代谢产物的神经毒性导致神经元及功能障碍 [14, 17]。
2014 年北京大学第一医院儿科曾对28例戊二酸尿症1型患者进行了研究,其中3例经新生儿筛查发现,25例患者于2个月至17岁获诊。22例(79%)为婴儿期起病,表现为智力、运动落后或倒退,肌张力障碍,癫癎发作,手足徐动,间断呕吐、喂养困难和嗜睡;其中20例遗留不同程度的智力、运动落后,仅2例发育正常。3例(11%)患者为晚发型,于8~16岁发病,临床表现包括运动倒退、阵发性头痛和癫癎发作,治疗后恢复良好,智力、运动恢复如常。23例患者接受了头颅核磁共振检查,其中22例有异常表现,包括特征性的外侧裂增宽、基底节损害、脑白质异常和脑萎缩[8, 15]。
3 生化表现戊二酸尿症1型患者常规实验室检查通常无特异性异常。急性发作期可有代谢性酸中毒、低血糖、酮症和高氨血症等表现,必须通过尿有机酸和血酯酰肉碱谱分析进行生化诊断[15]。患者血中戊二酰肉碱升高,常合并继发性肉碱缺乏症。尿液有机酸分析可见戊二酸和3-羟基戊二酸水平升高,3-羟基戊二酸为诊断性指标 [18]。根据尿戊二酸水平,可将戊二酸尿症1型患者分为高分泌型(>100 μmol/mol肌酐)和低分泌型(≤ 100 μmol/mol肌酐),低分泌型患者的尿戊二酸水平可正常。此外,患者尿二羧基酸与琥珀酸可轻度增高,提示线粒体能量代谢损害[19, 20]。
Sauer等在动物实验中发现,血液游离肉碱过低可导致相应的酯酰肉碱浓度下降[8, 15, 19]。戊二酰肉碱是戊二酰辅酶A和游离肉碱结合后的一种解毒排泄形式,能间接反映戊二酰辅酶A水平,如果游离肉碱过低,戊二酰肉碱生成减少,不能反映患者体内真实的代谢紊乱程度,导致假阴性结果[8, 15, 19]。
北京大学第一医院28例戊二酸尿症1型患儿的尿戊二酸、3-羟基戊二酸浓度均显著升高;22例患者治疗前接受了血液酯酰肉碱谱分析,其中1例血液戊二酰肉碱水平正常,考虑为严重的继发性肉碱缺乏导致的假阴性结果,经左卡尼汀治疗后血液戊二酰肉碱显著增高[8, 15]。建议对于临床疑似戊二酸尿症1型的患者,应及早进行尿液有机酸分析,动态监测血液酯酰肉碱谱,以明确诊断。
4 GCDH基因编码戊二酰辅酶A脱氢酶的基因GCDH突变是导致戊二酸尿症1型的遗传病因。GCDH位于染色体19p13.2,约7 kb,含11个外显子[21]。至今已报道约110种突变,包括错义突变、无义突变和内含子突变等。不同种族人群具有不同的热点突变。c.1240C>T为德国人群中报道较多的突变,位于外显子10,导致戊二酰辅酶A脱氢酶活性显著降低[22]。澳大利亚和西班牙人群中较为常见的突变分别为c.298C>T和c.913G>A[22, 23]。c.914C>T在日本患者中携带率为12.1%[24]。
北京大学第一医院对来自大陆不同地区28例患者进行研究,共检出GCDH基因35种不同类型的突变,其中3例纯合突变、25例复合杂合突变,包括30种错义突变、3种移码突变、1种无义突变和1种终止密码子突变,以c.148T>C最常见,发生率为7.7% [8, 15]。虽然IVS10-2A>C曾被报道在中国南方地区较为常见[25, 26],但28例患者中未检出IVS10-2A>C。既往研究显示,不同的GCDH突变导致的酶活性降低程度可能与患者生化表型有关[27]。但国内外研究均未发现GCDH基因型与患者临床表型的相关性,相同基因型的患病同胞临床表现可差异显著,不同基因型和生化表型的患者临床表现也可能非常相似[8, 15]。
5 戊二酸尿症1型的诊断戊二酸尿症1型患者发病年龄不一,可自生后数日至成年发病,临床症状多样,临床诊断困难[8, 15, 24]。对于大头畸形合并额颞叶萎缩、伴或不伴椎体外系症状的患者需注意戊二酸尿症1型的可能,应及早进行血液酯酰肉碱谱、尿液有机酸和GCDH基因分析,争取早期诊断[8, 15]。
脑MRI是评估戊二酸尿症1型患者脑损伤的重要手段,明显优于CT。脑深部灰质核团基底节对称性损害及中央区白质损害是戊二酸尿症1型的脑部MRI主要表现;额颞部脑萎缩、双侧侧裂池明显扩大为戊二酸尿症1型特征性MRI表现[13, 28]。但是,并非所有的戊二酸尿症1型均呈现典型的影像改变。北京大学第一医院28例患者中23例接受了头颅核磁共振检查,22例有异常表现,如特征性的外侧裂增宽、基底节损害、脑白质异常和脑萎缩,1例正常[8, 15]。
尿有机酸分析、血液酯酰肉碱谱分析是诊断戊二酸尿症1型的关键技术。患者尿液戊二酸、3-羟基戊二酸、血液戊二酰肉碱水平增高可诊断戊二酸尿症1型。但应警惕低分泌型患者,需酶活性检测或基因突变分析进一步鉴别(表 1)[15, 29, 30]。
|
|
表 1 3 种戊二酸尿症的酶缺陷、基因缺陷、生化及临床特点 |
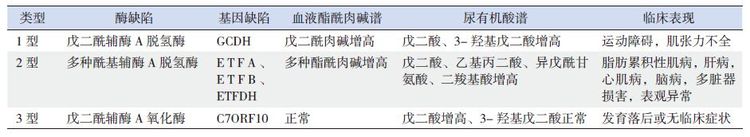
很多疾病可导致尿液戊二酸增高,需要进行鉴别诊断。进食含赖氨酸的食品或药品,尿液戊二酸轻度增高,但停用赖氨酸后尿戊二酸逐渐消失。另需与其他导致体内戊二酸增高的疾病鉴别,如多种酰基辅酶A脱氢酶缺陷,又被称为戊二酸尿症2型,是由于基因突变导致电子传递障碍引起线粒体多种脂肪酸和有机酸代谢障碍,导致脑、心肌、骨骼肌、肝脏等多脏器损害,急性期常合并低血糖、酸中毒、高脂血症、高尿酸血症[31, 32]。戊二酸尿症2型患者虽然尿戊二酸水平可增高,常合并二羧基酸尿症,乙基丙二酸、2-羟基戊二酸、异戊酰甘氨酸水平亦增高。戊二酸尿症2型患者血液酯酰肉碱谱分析可见多种酰基肉碱水平增高[32]。ETFA、ETFB、ETFDH基因突变分析可协助确诊(表 1)[33]。C7ORF10基因突变导致过氧化物酶中戊二酰辅酶A氧化酶缺陷,尿戊二酸增高,被称为戊二酸尿症3型[34, 35],目前报道较少,其临床表现不一,可无临床症状或表现出发育落后,尿戊二酸水平显著增高,但3-羟基戊二酸水平正常;血液酯酰肉碱结果正常(表 1)[35]。随着对戊二酸产生机制研究的进展,戊二酸尿症的疾病命名将以酶缺陷形式命名。
7 新生儿筛查和产前诊断欧美、日本、台湾等地区已经将戊二酸尿症1型纳入了新生儿筛查的疾病范围之内,我国上海、杭州、广州也进行了大样本量的研究[4]。血液戊二酰肉碱是筛查戊二酸尿症1型的主要指标,但低分泌型患者其血中戊二酰肉碱水平可能正常,致使筛查阴性,导致漏诊[8, 15, 36]。对于血液游离肉碱显著降低的新生儿应同时检测尿液酯酰肉碱谱及尿有机酸,提高筛查的检出率[8, 15, 37]。对于新生儿筛查结果阳性的患者还需通过尿液有机酸分析、酶学检测或基因分析进一步明确诊断。戊二酸尿症1型的早期诊断和治疗可改善预后,避免或减轻神经系统损害[38]。
对于曾经生育戊二酸尿症1型患者的母亲,再孕时可在妊娠中期进行产前诊断。自上纪80年代,羊水有机酸分析就已应用于戊二酸尿症1型的产前诊断[39]。但是,低分泌型的戊二酸尿症1型患儿尿液戊二酸正常,仅通过羊水有机酸分析可能出现漏诊。在先证者GCDH突变明确的基础上,通过胎盘绒毛细胞或羊水细胞GCDH突变分析可进行胎儿产前诊断,国内外已获得成功的经验[40]。
8 治疗与预后戊二酸尿症1型的治疗包括长期维持治疗和应急治疗。维持治疗主要为补充左卡尼汀及饮食干预。饮食治疗原则为限制赖氨酸及色氨酸,并补充无赖氨酸和低色氨酸的特殊氨基酸配方,将患者血赖氨酸浓度控制在60~120 μmol/L[41, 42]。为保证生长发育,需要个体化饮食指导,保证足够的热卡、维生素、矿物质及微量元素的供应。动物实验证实限制赖氨酸饮食治疗可降低戊二酸和3-羟基戊二酸在脑组织中的沉积[43]。为保证脑发育,婴幼儿时期需要严格的饮食干预,6岁后可适当放宽饮食限制、不用限赖氨酸饮食,但是需注意不要过多摄入天然蛋白质,最好选择赖氨酸含量较低的食物[42]。维持期左卡尼汀剂量一般为每天50~100 mg/kg,根据血液游离肉碱水平、一般状况等调整剂量,将血浆游离肉碱维持在50~90 μmol/L,需终身维持治疗[15, 38]。此外,精氨酸可降低赖氨酸在脑内的沉积和氧化,补充精氨酸对戊二酸尿症1型患者有神经保护作用[45, 46]。在维持治疗中,不建议使用维生素B2、抗癫癎药物、谷氨酸受体拮抗剂、肌酸和抗氧化剂[42]。对于肢体僵硬、运动障碍的患者,可添加巴氯芬(每天1~2 mg/kg)或地西泮(每天0.1~1 mg/kg)。盐酸苯海索常用于治疗肌张力不全[15, 42]。
应急治疗是预防急性纹状体病变的关键[42, 47]。婴幼儿期是病毒及细菌感染高发的年龄,容易合并急性获得性疾病,如发热、腹泻、呕吐、惊厥等,需尽快给予应急治疗。应急治疗应遵循自身中毒类型的代谢性疾病的基本治疗原则,积极祛除诱因,提供足够能量供应,增加左卡尼汀摄入量,降低天然蛋白摄入,并维持正常的体液、电解质和酸碱状态[47]。在应急治疗期间,患者每日能量摄入为同年龄正常需要量的120%,高碳水化合物饮食(如谷物、水果、根茎类蔬菜),停止摄入天然蛋白质,维持特殊氨基酸配方,每天静点葡萄糖15~20 g/kg,如果出现持续的血糖升高和尿糖阳性,可加用胰岛素每小时0.05 IE/kg。左卡尼汀每天100~200 mg/kg,有效促进有机酸排泄。若患者出现明显酸中毒,需静点碳酸氢钠。此外,根据不同临床症状,可酌情使用解热镇痛药、抗生素、利尿剂、抗癫癎药物[42, 47]。
6 岁以后患儿机体抗分解代谢能力增强,但在发热、手术、暴饮暴食和接种疫苗的情况下,仍有可能发生急性神经系统损伤,在这些情况下需密切观察意识状态,有无呕吐、高热或神经系统异常症状,及时进行应急治疗。
戊二酸尿症1型为小分子代谢病,药物及饮食治疗已取得成熟的经验。但戊二酰辅酶 A脱氢酶在脑、肝、肾、皮肤等多种组织表达,不适于进行细胞移植;而关于基因治疗亦尚待探讨,国内外尚未获得成熟的经验。
戊二酸尿症1型患者的预后与发病年龄、治疗时间有关,早期诊断及合理治疗可明显改善其预后。发病越早的患者预后越差[8, 15, 24]。研究证实,戊二酸尿症1型急性纹状体损伤多发生于2岁以前,通过新生儿筛查在症状前开始治疗的可降低神经系统致残率[1, 11, 48],而在纹状体损伤出现后获诊的患者大都遗留不同程度的神经系统损害[11]。国内外经验均证实了新生儿筛查的社会效益及经济效益[4, 48, 49]。
9 结语戊二酸尿症1型是一种常染色体隐性遗传代谢病,临床表现多样,可造成不可逆性脑损伤,早期诊断和合理治疗对本病尤为重要。对于临床疑似病例,尿有机酸分析、血液酯酰肉碱谱分析是诊断戊二酸尿症1型的关键技术,基因检测和酶学分析可确诊疾病。新生儿筛查是早期诊断和改善患者预后的最佳途径,戊二酸尿症1型新生儿筛查在全国普及十分必要。
| [1] | Lee CS, Chien YH, Peng SF, et al. Promising outcomes in glutaric aciduria type I patients detected by newborn screening[J]. Metab Brain Disease, 2013, 28(1): 61-67. |
| [2] | Schmiesing J, Schlüter H, Ullrich K, et al. Interaction of glutaric aciduria type 1-related glutaryl-CoA dehydrogenase with mitochondrial matrix proteins[J]. PLoS One, 2014, 9(2): e87715. |
| [3] | Kyllerman M, Steen G. Glutaric aciduria. A "common" metabolic disorder[J] Arch Fr Pediatr, 1980, 37(4): 279. |
| [4] | Niu DM, Chien YH, Chiang CC, et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan[J]. J Inherit Metab Dis, 2010, 33(Suppl 2): S295-S305. |
| [5] | Pierson TM, Nezhad M, Tremblay MA, et al. Adultonset glutaric aciduria type I presenting with white matter abnormalities and subependymal nodules[J]. Neurogenetics, 2015, 16(4): 325-328. |
| [6] | Young-Lin N, Shalev S, Glenn OA, et al. Teaching neuroimages: infant with glutaric aciduria type 1 presenting with infantile spasms and hypsarrhythmia[J]. Neurology, 2013, 81(24): e182-183. |
| [7] | Yang L, Yin H, Yang R, et al. Diagnosis, treatment and outcome of glutaric aciduria type I in Zhejiang Province, China[J]. Med Sci Monit, 2011, 17(7): 55-59. |
| [8] | Wang Q, Li X, Ding Y, et al. Clinical and mutational spectra of 23 Chinese patients with glutaric aciduria type 1[J]. Brain Dev, 2014, 36(9): 813-822. |
| [9] | Yang Y, Sujan S, Sun F, et al. Acute Metabolic Crisis Induced By Vaccination in Seven Chinese Patients[J]. Pediatric neurology, 2006, 35(2): 114-118. |
| [10] | Boy N, Heringer J, Haege G, et al. A cross-sectional controlled developmental study of neuropsychological functions in patients with glutaric aciduria type Ⅰ[J]. Orphanet J Rare Dis, 2015, 10(1): 163. |
| [11] | Kamate M, Patil V, Chetal V, et al. Glutaric aciduria type I: A treatable neurometabolic disorder[J]. Ann Indian Acad Neurol, 2012, 15(1): 31-34. |
| [12] | 陈靖, 王朝霞, 张锦丽, 等. 八例戊二酸尿症I 型患者的 GCDH 基因突变分析 [J]. 中华医学遗传学杂志, 2011, 28(4): 374-378. |
| [13] | Nunes J, Loureiro S, Carvalho S, et al. Brain MRI findings as an important diagnostic clue in glutaric aciduria type 1[J]. Neuroradiol J, 2013, 26(2):155-161. |
| [14] | Mohammad SA, Abdelkhalek HS, Ahmed KA , et al. Glutaric aciduria type 1: neuroimaging features with clinical correlation[J]. Pediatr Radiol , 2015, 45(11): 1696-1705. |
| [15] | 王峤, 丁圆, 刘玉鹏, 等. 戊二酸尿症1 型28 例的临床与实验室特征 [J]. 中华儿科杂志, 2014, 52(6): 415-419. |
| [16] | Harting I, Boy N, Heringer J, Seitz A, et al.1H-MRS in glutaric aciduria type 1: impact of biochemical phenotype and age on the cerebral accumulation of neurotoxic metabolites[J]. J Inherit Metab Dis, 2015, 38(5): 829-838. |
| [17] | Kurtcan S, Aksu B, Alkan A, et al. MRS features during encephalopathic crisis period in 11 years old case with GA-1[J]. Brain Dev, 2015, 37(5): 546-551. |
| [18] | Al-Dirbashi OY, Kölker S, Ng D, et al. Diagnosis of glutaric aciduria type 1 by measuring 3-hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry[J]. J Inherit Metab Dis, 2011, 34(1): 173-180. |
| [19] | Sauer SW, Opp S, Hoffmann GF, et al. Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I[J]. Brain, 2011, 134(Pt 1): 157-170. |
| [20] | Kölker S, Christensen E, Leonard JV, et al. Diagnosis and management of glutaric aciduria type I -revised recommendations[J]. J Inherit Metab Dis, 2011, 34(3): 677-694. |
| [21] | Marti-Masso JF, Ruiz-Martínez J, Makarov V, et al. Exome sequencing identifies GCDH(glutaryl-CoA dehydrogenase) mutations as a cause of a progressive form of early-onset generalized dystonia[J]. Hum Genet, 2012, 131(3): 435-442. |
| [22] | Zschocke J, Quak E, Guldberg P, et al. Mutation analysis in glutaric aciduria type I [J]. J Med Genet, 2000, 37(3): 177-181. |
| [23] | Busquets C, Merinero B, Christensen E, et al. Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically, and biochemically distinct[J]. Pediatr Res, 2000, 48(3): 315-322. |
| [24] | Mushimoto Y, Fukuda S, Hasegawa Y, et al. Clinical and molecular investigation of 19 Japanese cases of glutaric acidemia type 1[J]. Mol Genet Metab, 2011, 102(3): 343-348. |
| [25] | Tang NL, Hui J, Law LK, et al. Recurrent and novel mutations of GCDH gene in Chinese glutaric acidemia type I families[J]. Hum Mutat, 2000, 16(5): 446. |
| [26] | Lin SK, Hsu SG, Ho ES, et al. Novel mutation and prenatal sonographic findings of glutaric aciduria(type I) in two Taiwanese families[J]. Prenat Diagn, 2002, 22(8): 725-729. |
| [27] | Keyser B, Mühlhausen C, Dickmanns A, et al. Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH) [J]. Hum Mol Genet, 2008, 17(24): 3854-3863. |
| [28] | 李志勇, 干芸根, 方佃刚, 等. 戊二酸尿症I 型患者的临床与脑部MRI 特征 [J]. 中国CT 和MRI 杂志, 2014, 12(2): 22-24. |
| [29] | Pineda M, Ribes A, Busquets C, et al. Glutaric aciduria type I with high residual glutaryl-CoA dehydrogenase activity[J]. Dev Med Child Neurol, 1998, 40(12): 840-842. |
| [30] | Mushimoto Y, Hasegawa Y, Kobayashi H, et al. Enzymatic evaluation of glutaric acidemia type 1 by an in vitro probe assay of acylcarnitine profiling using fibroblasts and electrospray ionization/tandem mass spectrometry(MS/MS) [J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2009, 877(25): 2648-2651. |
| [31] | Zhu M, Zhu X, Qi X, et al. Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in mainland Chinese patients[J]. J Hum Genet, 2014, 59(5): 256-261. |
| [32] | Frerman FE, Goodman SI. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric aciduria type 2 [M]// Scriver CR, Beaudet AL, Sly WS, et al.The metabolic & molecular basis of inherited disease. 8th ed. New York: McGraw-Hill, 2001, 2357-2365. |
| [33] | Grünert SC. Clinical and genetical heterogeneity of lateonset multiple acyl-coenzyme A dehydrogenase deficiency[J]. Orphanet J Rare Dis, 2014, 9: 117. |
| [34] | Marlaire S, Van Schaftingen E, Veiga-da-Cunha M. C7orf10 encodes succinate-hydroxymethylglutarate CoA-transferase, the enzyme that converts glutarate to glutaryl-CoA[J]. J Inherit Metab Dis, 2014, 37(1): 13-19. |
| [35] | Sherman EA, Strauss KA, Tortorelli S, et al. Genetic Mapping of Glutaric Aciduria, Type 3, to Chromosome 7 and Identification of Mutations in C7orf10[J]. Am J Hum Genet, 2008, 83(5): 604-609. |
| [36] | Moore T, Le A, Cowan TM. An improved LC-MS/MS method for the detection of classic and low excretor glutaric acidemia type 1[J]. J Inherit Metab Dis, 2012, 35 (3): 431-435. |
| [37] | Jiang M, Liu L, Mei H, et al. Detection of inborn errors of metabolism using GC-MS: over 3 years of experience in southern China[J]. J Pediatr Endocrinol Metab, 2015, 28(3-4): 375-380. |
| [38] | Afroze B, Yunus ZM. Glutaric aciduria type 1--importance of early diagnosis and treatment [J]. J Pak Med Assoc, 2014, 64(5): 593-595. |
| [39] | Chalmers RA, Cheng KN, English NR, et al. Glutaric aciduria type I: prenatal exclusion using GC-MS analysis of amniotic fluid and enzymology with oxidation of [6-14C]lysine[J]. J Inherit Metab Dis, 1989, 12(3): 335-336. |
| [40] | Radha Rama Devi A, Ramesh VA, Nagarajaram HA, et al. Spectrum of mutations in Glutaryl-CoA dehydrogenase gene in glutaric aciduria type I -Study from South India[J]. Brain Dev, 2016, 38(1): 54-60. |
| [41] | Couce ML, López-Suárez O, Bóveda MD, et al. Glutaric aciduria type I: outcome of patients with early-versus latediagnosis[ J]. Eur J Paediatr Neurol, 2013, 17 (4): 383-389. |
| [42] | Heringer J, Boy SP, Ensenauer R, et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I [J]. Ann Neurol, 2010, 68(5): 743-752. |
| [43] | Posset R, Opp S, Struys EA, et al. Understanding cerebral L-lysine metabolism: the role of L-pipecolate metabolism in Gcdh-deficient mice as a model for glutaric aciduria type I[J]. J Inherit Metab Dis, 2015, 38(2): 265-272. |
| [44] | Viau K, Ernst SL, Vanzo RJ, et al. Glutaric acidemia type 1: outcomes before and after expanded newborn screening[J]. Mol Genet Metab, 2012, 106(4): 430-438. |
| [45] | Kölker S, Boy SP, Heringer J, et al. Complementary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I -A decade of experience[J]. Mol Genet Metab, 2012, 107(1-2): 72-80. |
| [46] | Fu X, Gao H, Tian F, et al. Mechanistic effects of amino acids and glucose in a novel glutaric aciduria type 1 cell model[J]. PLoS One, 2014, 9(10): e110181. |
| [47] | Kölker S, Greenberg CR, Lindner M, et al. Emergency treatment in glutaryl-CoA dehydrogenase deficiency[J]. J Inherit Metab Dis, 2004, 27(6): 893-902. |
| [48] | Brown A, Crowe L, Beauchamp MH, et al. Neurodevelopmental profiles of children with glutaric aciduria type I diagnosed by newborn screening: a follow-up case series[J]. JIMD Rep, 2015, 18: 125-134. |
| [49] | Pfeil J, Listl S, Hoffmann GF, et al. Newborn screening by tandem mass spectrometry for glutaric aciduria type 1: a costeffectiveness analysis[J]. Orphanet J Rare Dis, 2013, 8: 167-177. |