 2016, Vol. 18
2016, Vol. 18



2. 暨南大学附属第一医院 中心实验室, 广东 广州 510632
线粒体DNA耗竭综合征(mitochondrial DNAdepletion syndrome,MDS)是一组因线粒体DNA拷贝数减少而导致组织器官能量产生障碍的常染色体隐性遗传病[1-2]。该病临床表现复杂多样,根据临床特点可分为不同类型,分别与不同基因突变相关。如肌病型与TK2基因突变相关,脑肌病型多为SUCLA2、SUCLG1或RRM2B基因突变所致,神经胃肠道脑病型患者多存在TYMP基因突变,而DGUOK、POLG、C10orf2或MPV17基因突变多表现为肝脑型[2]。研究表明,DGUOK基因突变可见于14%~20%的MDS病例,被认为是MDS的主要致病基因,同时也是肝脑型MDS的最常见病因[3-6]。
DGUOK基因定位于染色体2p13,由7个外显子组成,编码含277个氨基酸残基的脱氧鸟苷激酶。该酶为嘌呤脱氧核糖核苷酸补救合成途径的限速酶,在保持线粒体三磷酸脱氧核糖核苷(deoxyribonucleoside triphosphate,dNTP)池的稳定性方面发挥重要作用[7]。DGUOK基因突变将造成dNTP池不平衡,影响线粒体DNA复制效率,最终产生线粒体耗竭。自2001年由Mandel等[3]首次报道DGUOK基因突变导致的肝脑型MDS以来,有关该基因的分子生物学研究在不同种族人群中陆续开展[8-9],目前已发现50余种突变类型,包括错义突变、无义突变、缺失突变、插入突变、剪接突变等[6, 8-10]。目前国内关于MDS患儿诊治的文献报道还相当有限[11-12],临床诊治经验也需要进一步积累。本文报道1例MDS患儿的临床特征和DGUOK基因突变特点,希望能够作为引玉之砖,为本病诊治研究提供参考。
1 资料与方法 1.1 研究对象患儿,女,7月20 d,因皮肤巩膜黄染3月余入院。3月前无明显诱因下出现皮肤巩膜黄染,于当地市人民医院就诊,发现肝脾肿大,伴肝功能明显异常(表 1),即转入广州某医院就诊,以“婴儿肝炎综合征”收入院。期间查尿气相色谱-质谱分析显示半乳糖、半乳糖酸、4-羟基苯乳酸、4-羟基苯丙酮酸增高;血串联质谱分析发现棕榈酸酰基肉碱升高;肝脏CT检查显示肝脏增大伴密度降低,但未发现占位性病变;放射性核素动态显像示肝脏摄取、排泄功能明显受损。给予更换无乳糖奶粉、补充维生素及凝血因子、肝泰乐护肝等治疗2月余,肝脾进行性增大,肝功能无好转,并出现眼球震颤症状,遂转到我院儿科就诊。患儿自起病以来,喂养困难,睡眠欠佳,小便色黄,大便未发现异常。
| 表 1 患儿实验室指标随月龄增加的纵向比较 |

患儿系第1胎第1产,孕39周因胎位不正剖宫产出生,出生体重2.2 kg(<-3 SD),身长49.0 cm(正常),否认窒息复苏史,父母体健非近亲结婚,否认家族遗传病及传染病史。患儿自出生后体格及精神运动发育严重落后于同龄儿,7.5月仍竖头不稳,双手不能抓握,不能逗笑,应人能力差。
入院体检:体重4.0 kg(<-3 SD),身长60.0 cm(<-3 SD),头围37.5 cm(<-3 SD)。神清,皮肤、巩膜黄染,未见皮疹、肝掌及蜘蛛痣。前囟平软,大小约0.5 cm×1.0 cm。双侧眼球水平震颤,瞳孔双侧等大等圆,对光反射灵敏。双肺呼吸音清,未闻及干湿罗音,心音有力,心率120次/min,各瓣膜听诊区未闻及病理性杂音。腹软,肝右肋下7.5 cm,质硬,脾左肋下3.0 cm,质中。四肢肌张力低,竖头不稳,不能翻身及坐立,双手不能主动抓物,腹壁、膝腱和跟腱等生理反射存在,克氏、布氏和巴士征均阴性。
辅助检查:血常规检查发现血红蛋白85 g/L(参考值110~120 g/L)。生化检查发现肝功能明显异常(表 1),血糖0.7 mmol/L(参考值2.2~7.0 mmol/L),血清锌3.2 μmol/L(参考值7.65~25.5 μmol/L)。凝血功能检查发现活化部分凝血活酶时间49.5 s(参考值28~44 s),凝血酶原时间16.9 s(11~16 s),凝血酶时间19.7 s(14~21 s),纤维蛋白原1.09 g/L(2~4 g/L)。25-羟维生素D3 6.07 ng/mL(30~100 ng/mL),血清铁蛋白345 ng/mL(4.6~204 ng/mL)。SLC25A13基因高频突变筛查未发现c.851del4,c.1638-1660dup,IVS6+5G>A,IVS16ins3kb及IVS4ins6kb等突变。
1.2 外显子组捕获测序分析采集患儿静脉EDTA抗凝外周血2 mL按照Blood DNA Mini kit试剂盒(Simgen公司,中国)说明书步骤提取基因组DNA 3~5 μg,并将其打断扩增,建立含有与胆汁淤积性肝病相关基因(GALT、GALE、FAH、TAT、HPD、GSTZ1、SLC25A13及DGUOK等共233个基因)的全基因组文库。用液相捕获试剂盒(迈基诺公司,中国)捕获上述目标基因,然后利用新一代测序仪IlluminaHiSeq2000(Illumina公司,美国)进行高通量测序。测序平均深度不小于200×,得出数据后进行基因序列生物信息学分析,找出致病基因。
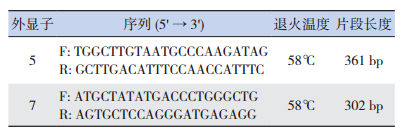
1.3 Sanger测序验证突变根据外显子组捕获测序检测结果,对患儿及其父母DNA标本进行DGUOK突变基因Sanger测序验证。用Blood DNA Mini kit试剂盒(Simgen公司,中国)提取该家系基因组DNA,用PCR试剂盒(Finnzymes Oy公司,芬兰)扩增突变所在外显子序列。所用引物(表 2)根据DGUOK基因的DNA序列使用Primer Premier 5.0软件设计,由英潍捷基贸易有限公司合成。聚合酶链反应体系为:94℃预变性5 min;然后94℃变性30 s,58℃退火40 s,72℃延伸50 s,共反应35个循环;最后再72℃延伸10 min。聚合酶链反应产物经琼脂糖凝胶电泳切胶纯化,由英潍捷基贸易有限公司测序。应用Chromas软件对测序结果进行分析,并应用DNAMAN软件与DGUOK基因组参考序列进行比对(Ensemble genome browser: ENST00000264093),突变的命名参考相关文献[13-14]。
| 表 2 Sanger测序验证DGUOK基因突变位点所在DNA序列扩增和测序引物 |
1.4 正常蛋白和突变蛋白三级结构对比分析
用SWISS-MODEL软件(http://swissmodel.expasy.org/)对野生型和突变型脱氧鸟苷激酶蛋白结构进行预测和比较,判断DGUOK基因突变对编码蛋白的影响,从而推断新突变的致病性。
本研究经医院医学伦理委员会批准并获得患儿父母知情同意。
2 结果 2.1 遗传学分析结果外显子组捕获测序未发现FAH、TAT、HPD和GSTZ1等酪氨酸血症致病基因突变;半乳糖血症相关基因GALT和GALE,以及Citrin缺陷病相关基因SLC25A13也未检测到致病性突变。但该技术在患儿DGUOK基因中检测到2个突变,一个为位于外显子5的错义突变c.679G>A,另一个为位于外显子7的移码突变c.817delT(图 1)。Sanger测序验证也确认该患儿为突变c.679G>A和c.817delT的复合杂合子,两个突变分别来自于患儿母亲和父亲(图 2)。其中c.679G>A为已报到致病性突变,而突变c.817delT在千人基因组数据库、亚洲本地正常人数据库、欧裔及非裔美国正常人数据库中均未检测到。经检索中国知网、万方、维普和美国Pubmed等国内外文献数据库,证实为未报道过的新突变。该新突变c.817delT造成移码,致使273位氨基酸由苯丙氨酸变为亮氨酸,随之遇到终止密码子,使翻译提前终止,理论上产生一个截短蛋白p.F273Lfs274X。

|
图 1 患儿外显子捕获测序截图 左图:该患儿DGUOK基因编码序列679位碱基(箭头所示)除了正常的鸟嘌呤(G)外,还有部分测序结果为腺嘌呤(A);右图:患儿DGUOK基因编码序列817位碱基(箭头所示)野生型为T,部分测序结果显示该碱基缺失。外显子捕获测序结果提示该患儿系DGUOK基因突变c.679G>A和c.817delT的复合杂合子。 |

|
图 2 患儿及其父母DGUOK基因Sanger测序图 A:患儿系c.679G>A和c.817delT的复合杂合子;B:父亲为c.817delT突变携带者;C:母亲为c.679G>A突变携带者。箭头所示为突变位点。 |
2.2 蛋白结构预测
用SWISS-MODEL分别构建野生型和突变型脱氧鸟苷激酶蛋白3D模型,分析突变c.817delT对脱氧鸟苷激酶结构造成的影响。结果显示,突变c.817delT致使脱氧鸟苷激酶二聚体C端第9个α-螺旋末端部分缺失,影响该酶底物磷酸盐供体与酶蛋白的结合,见图 3。

|
图 3 野生和突变型脱氧鸟苷激酶二聚体蛋白质结构飘带图 左图:显示野生型脱氧鸟苷激酶二聚体,可见位于C端的第9个α-螺旋结构完整,底物磷酸盐供体可与第9个α-螺旋正常结合(白色方框);右图:显示c.817delT突变发生后脱氧鸟苷激酶二聚体C端第9个α-螺旋末端部分缺失,导致底物磷酸盐供体无法结合(黄色方框)。 |
2.3 治疗与结局
患儿入院后3 d出现发热、咳嗽、流涕等呼吸道感染症状。住院期间给予护肝、输注血浆补充凝血因子,浓缩红细胞纠正贫血,补充维生素D和锌、纠正低血糖及抗感染等治疗,住院19 d,感染控制后应家属要求出院。出院诊断“肝硬化;重度营养不良;支气管炎;贫血;维生素D缺乏症;锌缺乏症”。1月余后在家中死亡。死亡后1年半,最终通过外显子组捕获测序及Sanger测序验证,确诊DGUOK基因突变导致的MDS。
3 讨论DGUOK基因突变导致的肝脑型MDS临床表现特异性不强,因此对怀疑本病患儿进行分子遗传学分析十分重要。DGUOK基因编码的脱氧鸟苷激酶是一个同源二聚体蛋白,每一个亚基包含5个β-片层和9个α-螺旋结构,其中第9个α-螺旋可与底物磷酸盐供体结合,对该酶催化作用至关重要[15-16]。本研究通过外显子组捕获测序及一代测序验证,证实患儿为DGUOK基因突变c.679G>A和c.817delT的复合杂合子。前者来自于母亲,为已报道的致病突变[4, 17];后者系父源性移码突变,国内外尚未见文献报道。SWISS-MODEL模型预测结果表明,DGUOK基因突变c.817delT使得其编码的脱氧鸟苷激酶蛋白质翻译提前终止,影响C端第9个α-螺旋的结构完整性,导致磷酸盐供体结合障碍,从而影响脱氧鸟苷激酶的催化作用。两个突变均影响DGUOK基因编码产物脱氧鸟苷激酶的功能,造成dNTP池不平衡,影响线粒体DNA复制效率,最终产生线粒体耗竭,致使患儿出现MDS的一系列临床表现。
本文MDS患儿主要表现为肝脾肿大和肝功能受损,同时具有精神运动发育迟滞、眼球震颤及肌张力减低等神经系统异常表现,为比较典型的肝脑型MDS患儿。上述表现与文献报道极为相似[3-6, 9, 18]。细胞质内存在脱氧核糖核苷激酶营救效应,可部分代偿DGUOK基因突变后细胞质内失活的脱氧鸟苷激酶[19]。但是,人类大脑和肝脏中脱氧鸟苷激酶和脱氧核糖核苷激酶的活性均较低[19-20],这可能是本研究MDS患儿以肝脑表现为主要临床特点的原因。
实验室检查方面,本研究患儿甲胎蛋白升高显著,低血糖和高血清铁蛋白特点也较突出。有学者认为血中甲胎蛋白升高是DGUOK基因相关肝脑型MDS的主要生化指标[3, 6]。甲胎蛋白升高与受损肝细胞的代偿再生有关。Scheers等[21]报道呼吸链疾病甲胎蛋白升高与肝脏肿瘤相关。有文献报道在DGUOK疾病中有2例甲胎蛋白水平分别达到39 634.9 ng/mL和10 961.5 ng/mL,发生了肝脏的恶性肿瘤[22]。本研究患儿甲胎蛋白最高升至431 632 ng/mL,但肝脏CT检查未发现占位性病变。绝大部分报道DGUOK相关肝脑型MDS都伴随严重的低血糖[3, 4, 6, 8, 10]。Sezer等[6]及Pronicka等[10]均发现DGUOK相关MDS患儿中有胰岛细胞增生现象。MDS时低血糖对肝功能损害更严重[10]。本研究患儿血糖低至0.7 mmol/L,反应差,出虚汗,未见有呼吸困难、青紫、颤抖及惊厥表现。给予葡萄糖纠正低血糖,血糖维持2.88~3.39 mmol/L。该患儿低血糖是否与胰岛细胞增生有关,尚需进一步研究。DGUOK肝脑型MDS患儿中发现铁过载伴转铁蛋白及铁蛋白升高,同时在肝细胞及枯否细胞中发现铁集聚[10, 23]。DGUOK基因突变加重铁过载对肝细胞的损伤[10],铁过载同时影响了线粒体DNA水平,有文献报道在铁沉积症的患儿发生严重线粒体DNA耗竭(线粒体DNA只有正常水平的9%)[24]。
DGUOK基因突变导致的MDS临床病死率高,患儿多在6个月前发病,1岁之前死亡[3, 6]。本病目前缺乏有效的治疗方式,虽有使用维生素、呼吸底物及辅酶治疗本病患儿的报道,但疗效甚微[7]。本文患儿诊治经过及其不良结局,符合这一特点。本病仅肝脏受累而无神经系统异常者可选择肝脏移植,当患儿出现精神运动发育迟滞或眼球震颤时,肝脏移植效果不佳[23, 25-26]。
总之,本研究系统分析了1例肝脑型MDS患儿的临床症状、体征和实验室检查特点,并通过外显子组捕获测序及Sanger测序验证,确诊患儿为DGUOK基因突变导致的MDS,且发现1个新突变c.817delT。本研究扩展了DGUOK基因突变谱,并为患者分子诊断、家系遗传咨询及产前诊断提供了实验依据。
| [1] | Spinazzola A, Invernizzi F, Carrara F, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes[J]. J Inherit Metab Dis,2009, 32 (2) :143–158 . |
| [2] | El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options[J]. Neurotherapeutics,2013, 10 (2) :186–198 . |
| [3] | Mandel H, Szargel R, Labay V, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA[J]. Nat Genet,2001, 29 (3) :337–341 . |
| [4] | Salviati L, Sacconi S, Mancuso M, et al. Mitochondrial DNA depletion and dGK gene mutations[J]. Ann Neurol,2002, 52 (3) :311–317 . |
| [5] | Ji JQ, Dimmock D, Tang LY, et al. A novel c.592-4_c.592-3delTT mutation in DGUOK gene causes exon skipping[J]. Mitochondrion,2010, 10 (2) :188–191 . |
| [6] | Sezer T, Ozçay F, Balci O, et al. Novel deoxyguanosine kinase gene mutations in the hepatocerebral form of mitochondrial DNA depletion syndrome[J]. J Child Neurol,2015, 30 (1) :124–128 . |
| [7] | Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes--many genes, common mechanisms[J]. Neuromuscul Disord,2010, 20 (7) :429–437 . |
| [8] | Dimmock DP, Zhang Q, Dionisi-Vici C, et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase[J]. Hum Mutat,2008, 29 (2) :330–331 . |
| [9] | Yamazaki T, Murayama K, Compton AG, et al. Molecular diagnosis of mitochondrial respiratory chain disorders in Japan: focusing on mitochondrial DNA depletion syndrome[J]. Pediatr Int,2014, 56 (2) :180–187 . |
| [10] | Pronicka E, Węglewska-Jurkiewicz A, Taybert J, et al. Post mortem identification of deoxyguanosine kinase (DGUOK) gene mutations combined with impaired glucose homeostasis and iron overload features in four infants with severe progressive liver failure[J]. J Appl Genet,2011, 52 (1) :61–66 . |
| [11] | 刘志梅, 方方, 丁昌红, 等. 二代测序在儿童线粒体病诊断中的应用[J]. 中华儿科杂志,2015,53 (10) :747–753. |
| [12] | 刘志梅, 方方, 丁昌红, 等. SUCLA2相关脑肌病型线粒体DNA耗竭综合征一例并文献复习[J]. 中华儿科杂志,2014,52 (11) :817–821. |
| [13] | den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations[J]. Hum Genet,2001, 109 (1) :121–124 . |
| [14] | den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion[J]. Hum Mutat,2000, 15 (1) :7–12 . |
| [15] | Johansson K, Ramaswamy S, Ljungcrantz C, et al. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases[J]. Nat Struct Biol,2001, 8 (7) :616–620 . |
| [16] | Eriksson S, Munch-Petersen B, Johansson K, et al. Structure and function of cellular deoxyribonucleoside kinases[J]. Cell Mol Life Sci,2002, 59 (8) :1327–1346 . |
| [17] | Lee NC, Dimmock D, Hwu WL, et al. Simultaneous detection of mitochondrial DNA depletion and single-exon deletion in the deoxyguanosine gene using array-based comparative genomic hybridisation[J]. Arch Dis Child,2009, 94 (1) :55–58 . |
| [18] | Kiliç M, Sivri HS, Dursun A, et al. A novel mutation in the DGUOK gene in a Turkish newborn with mitochondrial depletion syndrome[J]. Turk J Pediatr,2011, 53 (1) :79–82 . |
| [19] | Saada-Reisch A. Deoxyribonucleoside kinases in mitochondrial DNA depletion[J]. Nucleosides Nucleotides Nucleic Acids,2004, 23 (8-9) :1205–1215 . |
| [20] | Rötig A, Poulton J. Genetic causes of mitochondrial DNA depletion in humans[J]. Biochim Biophys Acta,2009, 1792 (12) :1103–1108 . |
| [21] | Scheers I, Bachy V, Stephenne X, et al. Risk of hepatocellular carcinoma in liver mitochondrial respiratory chain disorders[J]. J Pediatr,2005, 146 (3) :414–417 . |
| [22] | Freisinger P, Futterer N, Lankes E, et al. Hepatocerebral mitochondrial DNA depletion syndrome caused by deoxyguanosine kinase (DGUOK) mutations[J]. Arch Neurol,2006, 63 (8) :1129–1134 . |
| [23] | Labarthe F, Dobbelaere D, Devisme L, et al. Clinical, biochemical and morphological features of hepatocerebral syndrome with mitochondrial DNA depletion due to deoxyguanosine kinase deficiency[J]. J Hepatol,2005, 43 (2) :333–341 . |
| [24] | Vu TH, Tanji K, Holve SA, et al. Navajo neurohepatopathy: a mitochondrial DNA depletion syndrome?[J]. Hepatology,2001, 34 (1) :116–120 . |
| [25] | Al-Hussaini A, Faqeih E, El-Hattab AW, et al. Clinical and molecular characteristics of mitochondrial DNA depletion syndrome associated with neonatal cholestasis and liver failure[J]. J Pediatr,2014, 164 (3) :553–559 . |
| [26] | Dimmock DP, Dunn JK, Feigenbaum A, et al. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency[J]. Liver Transpl,2008, 14 (10) :1480–1485 . |