 2016, Vol. 18
2016, Vol. 18


范可尼贫血(Fanconi anemia,FA)是一种常染色体或X 连锁隐性遗传性疾病,主要临床表现为先天性发育异常、进行性骨髓衰竭和早期发生血液肿瘤或其他实体瘤[1]。FA 治疗的主要方法有口服药物(雄激素+/- 糖皮质激素)、支持治疗(输注血液制品)、异基因造血干细胞移植及基因治疗等。我国FA 的治疗仍以口服药物治疗为主,但临床分型相同的的FA 患者,即使治疗相同,其疗效亦存在差别,原因尚不完全清楚,为此我们初步分析了6 例口服雄激素+ 糖皮质激素治疗的FA患者临床资料及基因分型相关内容,探讨FA 临床进展与基因分型之间的关系。
1 资料与方法 1.1 研究对象及分组回顾性分析2013 年9 月至2015 年3 月就诊于我院,经临床表现、血常规、骨髓象、单细胞凝胶电泳实验、丝裂霉素C(mitomycin C,MMC)诱导的染色体断裂实验、二代测序确诊为FA,并且均为口服药物治疗的6 例患者。6 例患者均予口服康力龙(每次2 mg,每日1 次)+ 泼尼松(每次 5 mg,每日1 次),3 个月无效则停用。根据血常规,将6 例患者分为轻、中、重度3 组,均予门诊复诊及电话随访,中位随访时间54 个月(13~84个月),并根据患者治疗后3,6,9,12 个月的血常规进行疗效评价。有效:初诊与随访时均为轻度或随访时由重度转为轻、中度以及中度转为轻度。无效:疾病分组由轻度转为中、重度,或由中度转为重度或者死亡。
FA 的诊断及分组依据2008 年制定的FA 诊断及分组标准[2] 进行。
1.2 单细胞凝胶电泳检测彗星细胞率患者初诊时取静脉血5 mL(肝素抗凝),利用密度梯度离心法进行单个核细胞分离。调整细胞最终浓度至105~106/mL,取30 μL 细胞悬液进行琼脂糖凝胶电泳,计算彗星细胞率。实验具体步骤及彗星细胞率的计算方法参见文献[3-4]。彗星细胞率>30% 可诊断FA。
1.3 染色体断裂实验采用IAEA 的标准化方法[5] 进行染色体培养,24 h 后加入MMC,继续培养48 h,收获细胞,预固定、低渗制片,普通光镜观察,镜下计数100个分裂相,计数染色体畸变细胞,进行畸变率分析。如染色体畸变率>30% 为阳性,可诊断为FA。丝裂霉素C 的浓度选取0、40、80 ng/mL。每份标本分析100 个分裂相。
1.4 FA 基因测序利用基于第二代测序方法设计的包含249 种先天性骨髓衰竭性疾病的诊断试剂盒(北京迈基诺基因科技有限责任公司),对患者外周血提取的DNA 进行FA 突变基因检测。对于基因检测没有纯合突变或复合杂合突变、不能进行基因分型的FA 患者利用互补实验进行检测(由印第安纳大学医学院赫尔曼儿科研究中心完成)。
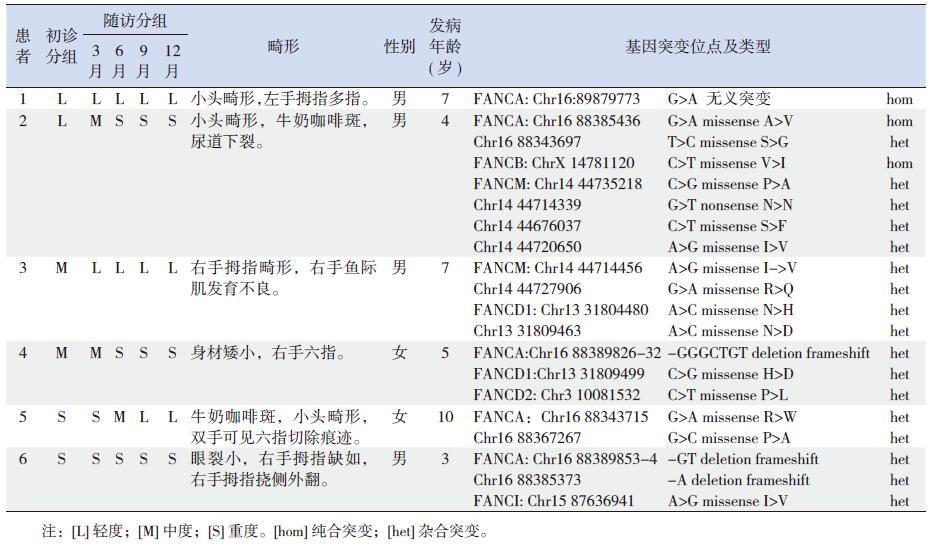
2 结果6 例患者的染色畸变率在32%~39%,彗星细胞率在39%~69% 之间,可诊断为FA。6 例患者中男性4 例、女性2 例,均有畸形;中位随访时间54 个月(13~84 个月)。6 例FA 患者中,5 例为FANCA 型、1 例为FANCM 型,4 例携带2 种及以上FA 基因突变。初诊时轻度、中度及重度各2 例,初诊为轻度的患者中,治疗12 个月随访时为轻度的患者1 为FANCA 基因的纯合突变,诊断为FANCA 型;治疗12 个月随访时为重度的患者2,除存在FANCA 的纯合突变、杂合突变外,还存在FANCB 的纯合突变、FANCM 的复合杂合突变,对其父母进行验证,显示患者母亲携带有FANCB 基因突变,对患者2 进行互补实验,确诊为FANCA 型。初诊为中度的两位患者中,治疗12个月随访为轻度的患者3 为FANCM 与FANCD1 的复合杂合突变,对其父母进行验证,显示患者的FANCM 两个突变一个来自母亲、一个来自父亲,提示患者为FANCM 型;而治疗12 个月随访时为重度的患者 4 却同时存在FANCA,FANCD1,FANCD2 三种基因突变,经过验证其母亲携带FANCA 基因的突变位点,虽然Chr1688389826-32delGGGCTGT 在Rockefeller 大学FA 突变数据库中未发现,但Chr1688389829-30delCT 在此数据库中报道过3 次,故此患者可诊断为FANCA 型。在初诊为重度的2 例患者中,治疗12 个月随访为轻度的患者5 为FANCA 的复合杂合突变,诊断为FANCA 型;而治疗12 个月随访为重度的患者6 除含有FANCA 突变外,还存在FANCI 的杂合突变,突变位点FANCA Chr16883853-54delGG在Rockefeller 大学FA 突变数据库中亦未发现,但Chr16 88385351-54delGAGG 报道过1 次,Chr1688385351delG reported 报道过3 次,根据相应结果分析,此患者为FANCA 型。见表 1。
| 表 1 6 例FA 患者的基因检测结果与临床特征 |
3 讨论
范可尼贫血(FA)是一种少见的常染色体或X 连锁隐性遗传性疾病,是最常见的遗传性骨髓衰竭综合征,其发病率为1~5/100 万,基因携带率为1/300[1]。FA 主要由相关基因突变所致,最终影响DNA 修复过程。根据突变基因种类的不同,目前FA 分为FANCA,FANCB,FANCC,FANCD1,FANCD2,FANCE,FANCF,FANCG,FANCI,FANCJ,FANCL,FANCM,FANCN,FANCO,FANCP,FANCQ 及FANCS 等17 种类型。不同类型的FA 患者其临床预后存在异质性,如FANCO与FANCS 患者到目前为止未见发展为骨髓衰竭性疾病的报道[6],故准确的基因分型对于FA 患者的预后判断尤为重要[7-9]。另外,不同的FA 基因杂合突变可产生不同的病理生理学作用,如FANCD1突变与乳腺癌的发生密切相关,FANCD2 突变与儿童T- 急性淋巴细胞白血病和睾丸精原细胞瘤有一定关系,单泛素化的FANCD2 与BCR-ABL1 激酶诱导的白血病发生相关等[10-17]。
国外报道的FA 基因突变多是同一个FA 基因的纯合或复合杂合突变,一个患者同时携带2 种或以上FA 突变基因的报道较少[18]。本研究6 例FA 患者中,5 例患者为FANCA 型,1 例患者为FANCM 型,4 例患者携带2 种及以上FA 基因突变。与国外研究不同,可能与FA 患者的遗传背景不同或FA 基因突变的携带率与国外存在差异,还需扩大样本量建立FA 突变数据库以进一步研究。本研究显示,治疗12 个月时评估为重度的3 例患者,发病年龄较小,均为2 种及以上的基因突变,提示不同的FA 基因突变类型临床转归可能不同,基因突变越复杂,患者疗效可能越差,越有可能进展为重型,此类患者应尽早进行造血干细胞移植。
本文通过临床资料与基因测序相结合,初步探讨了FA 临床转归与基因分型间的关系。但范可尼贫血是一种异质性较强的先天性骨髓衰竭性疾病,目前我国尚无FA 的流行病学及大样本数据分析,更没有FA 相关突变的数据库进行参考,还需进行多中心协作,建立FA 突变数据库,为FA 基因诊断应用于临床治疗的指导提供理论依据。
| [1] | D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway[J]. Nat Rev Cancer , 2003, 3 (1) : 23–34. DOI:10.1038/nrc970 |
| [2] | Blanche P.Diagnostic Evaluation of FA[M]//Mary EE, Dave F, Lynn F, et al. Fanconi anemia:Guidelines for diagnosis and management[M]. 3rd ed.Eugene: Fanconi Anemia Research Fund Inc, 2008 : 34 -53. |
| [3] | 张丽, 刘强, 邹尧, 等. 单细胞凝胶电泳试验在范可尼贫血诊断中的意义及其与丝裂霉素C试验相关性研究[J]. 中华儿科杂 , 2013, 51 (2) : 122–125. |
| [4] | 齐军元, 邵英起, 刘永泽, 等. 单个细胞凝胶电泳分析在范可尼贫血诊断中的应用[J]. 中华血液学杂志 , 2006, 27 (10) : 690–693. |
| [5] | International Atomic Energy Agency. Dicentric analysis[M]. Vienna: International Atomic Energy Agency, 2001 : 27 -59. |
| [6] | Wang AT, Smogorzewska A. SnapShot:Fanconi anemia and associated proteins[J]. Cell , 2015, 160 (1-2) : 354–354. DOI:10.1016/j.cell.2014.12.031 |
| [7] | Alan D, D'Andrea MD. The Fanconi anemia and breast cancer susceptibility pathway[J]. N Engl J Med , 2010, 362 (20) : 1901–1919. DOI:10.1056/NEJMoa0907006 |
| [8] | Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the international Fanconi anemia Registry (IFAR)[J]. Blood , 2003, 101 (4) : 1249–1256. DOI:10.1182/blood-2002-07-2170 |
| [9] | Myers K, Davies SM, Harris RE, et al. The clinical phenotype of children with Fanconi anemia caused by biallelic FANCD1/BRCA2 mutations[J]. Pediatr Blood Cancer , 2012, 58 (3) : 462–465. DOI:10.1002/pbc.v58.3 |
| [10] | Seal S, Thompson D, Renwick A, et al. Breast Cancer Susceptibility Collaboration (UK):Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles[J]. Nat Genet , 2006, 38 (11) : 1239–1241. DOI:10.1038/ng1902 |
| [11] | Rahman N, Seal S, Thompson D, et al. Breast Cancer Susceptibility Collaboration (UK):PALB2,which encodes a BRCA2-interacting protein,is a breast cancer susceptibility gene[J]. Nat Genet , 2007, 39 (2) : 165–167. DOI:10.1038/ng1959 |
| [12] | Reid S, Schindler D, Hanenberg H, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer[J]. Nat Genet , 2007, 39 (2) : 162–164. DOI:10.1038/ng1947 |
| [13] | Wark L, Novak D, Sabbaghian N, et al. Heterozygous mutations in the PALB2 hereditary breast cancer predisposition gene impact on the three-dimensional nuclear organization of patient-derived cell lines[J]. Genes Chromosomes Cancer , 2013, 52 (5) : 480–494. DOI:10.1002/gcc.v52.5 |
| [14] | Thompson ER, Doyle MA, Ryland GL, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles[J]. PLoS Genet , 2012, 8 (9) : el002894. |
| [15] | Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers[J]. Br J Cancer , 2007, 96 (1) : 11–15. DOI:10.1038/sj.bjc.6603535 |
| [16] | Smetsers S, Muter J, Bristow C, et al. Heterozygote FANCD2 mutations associated with childhood T cell ALL and testicular seminoma[J]. Fam Cancer , 2012, 11 (4) : 661–665. DOI:10.1007/s10689-012-9553-3 |
| [17] | Koptyra M, Stoklosa T, Hoser G, et al. Monoubiquitinated Fanconi anemia D2(FANCD2-Ub) is required for BCR-ABL1 kinase-induced leukemogenesis[J]. Leukemia , 2011, 25 (8) : 1259–1267. DOI:10.1038/leu.2011.91 |
| [18] | Chang L, Yuan W, Zeng H, et al. Whole exome sequencing reveals concomitant mutations of multiple FA genes in individual Fanconi anemia patients[J]. BMC Med Genomics , 2014, 7 : 24. DOI:10.1186/1755-8794-7-24 |