 2016, Vol. 18
2016, Vol. 18


进行性家族性肝内胆汁淤积症1 型(progressive familial intrahepatic cholestasis type Ⅰ,PFIC1),早期称Byler 氏病,是ATP8B1 基因突变引起的常染色体隐性遗传病[1-4]。ATP8B1 基因定位于染色体18q21-22,全长约176.68 kb,包含28 个外显子,编码含1 251 个氨基酸残基的FIC1 蛋白[3-4]。FIC1 蛋白主要分布于肠上皮细胞刷状缘、胃小凹上皮细胞和胰腺腺泡细胞顶端膜,以及肝细胞胆小管膜和胆管细胞顶端膜等处[4-8],在肾脏和内耳毛细胞也有表达[1-2]。FIC1 作为一种氨基磷脂易位酶,负责把磷脂酰丝氨酸从生物膜外层转移到内层,从而维持膜两侧脂质分布的不对称性[2]。PFIC1 患者通常在1 岁内出现进行性肝内胆汁淤积临床表现,可发展至终末期肝病;其最突出的生化特点是血清γ- 谷氨酰转肽酶(GGT)始终不高,与严重肝内胆汁淤积表现不平行[1-2]。由于FIC1 蛋白在多个系统均有表达,PFIC1 患者常伴多种肝外表现,如腹泻、胰腺炎、神经性耳聋等[2, 7]。
国外文献估计PFIC1 发病率大概为1/100 000至1/1 000 000,男女间无明显差异[3]。目前我国尚缺乏关于本病发病率的大样本多中心流行病学调查数据,确诊患儿局限于北京、上海、广州和武汉等中心城市的个别大型医院[8-11],不但患者例数相当有限,临床诊治经验也需进一步积累。本研究分析1 例PFIC1 患儿临床特点及其ATP8B1 基因突变特征,为本病后续诊治研究提供参考。
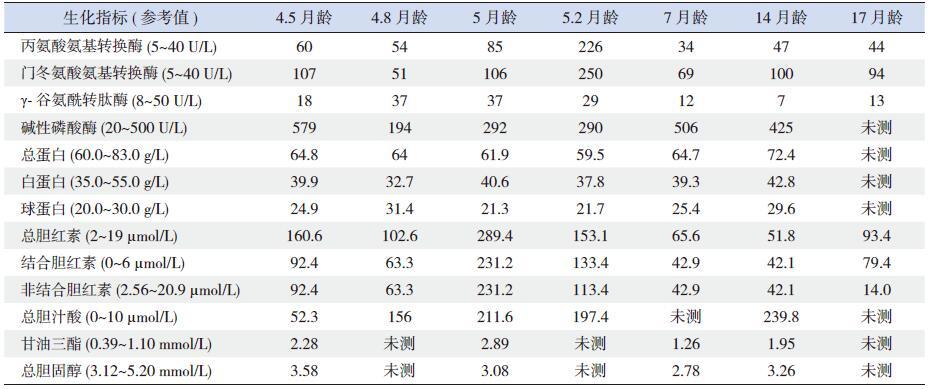
1 资料与方法 1.1 研究对象患儿,男,1 岁2 个月,因发现皮肤黄染10月余就诊。患儿4 月龄时,家长注意到其皮肤和巩膜黄染,但不伴白陶土样大便及茶色尿,未予重视。4.5 月龄时黄疸仍未消退,在当地医院查血生化发现肝功能异常(表 1),以婴儿肝炎综合征收住院,并予茵栀黄退黄、腺苷蛋氨酸护肝等治疗20 d,皮肤黄染逐渐加深,大便变浅黄。遂转至另一大型综合性医院儿科,体检除发现皮肤巩膜黄染外,肝脾均肿大,肝脏右肋下4 cm,剑突下2 cm,质韧,边锐,脾左肋下2 cm,质软。生化检查发现丙氨酸氨基转换酶(ALT)等肝功能指标明显异常。尿常规胆红素(3+)。大便常规和凝血功能检查未见异常。超声检查发现肝大,质地欠佳,胆总管未见局灶性扩张;脾、胆、胰、双肾无占位性。诊断考虑婴儿肝病综合征,予复方甘草酸苷、还原型谷胱甘肽保肝治疗半月余,黄疸、肝大和肝功能异常等均无明显改善。遂于6个月龄时行腹腔镜下胆道造影冲洗及肝活检微创手术,排除了胆道闭锁。肝脏病理见肝小叶结构凌乱,肝细胞弥漫性胆汁淤积伴毛细胆管扩张、胆栓形成;门管区胆管缺如,仅1 个门管区见有胆管,不伴胆汁淤积;网状纤维染色显示肝纤维化。出院后中西药结合治疗半年余,皮肤巩膜黄染进行性加重。为明确病因而至我院儿科就诊。
| 表 1 患儿历次生化检查结果 |
患儿自起病以来,精神反应一般,吃奶尚可,无呕吐,大便色浅,尿色深黄。
患儿系第1 胎第1 产,因胎位不正剖宫产出生,出生体重3 450 g,身长50 cm。出生时无缺氧窒息。患儿3 月龄抬头,生长发育与同龄人无明显差异。父母体健,非近亲结婚,否认遗传病家族史。
体格检查:体重10.5 kg,身长77 cm,头围46.5 cm。神清,反应可。皮肤黄染,颜面及躯干部为甚。头颅五官无畸形。前囟平软,大小约0.5 cm×0.5 cm。巩膜明显黄染,双侧瞳孔等大等圆,直径3 mm,对光反射灵敏。双肺呼吸音清,未闻及干湿罗音。心音有力,心率120 次/min,各瓣膜听诊区未闻及病理性杂音。腹部稍膨隆,软,肝右肋下4 cm,剑突下3 cm,质地中等,边缘锐利,脾左肋下2 cm 可触及,质软。四肢肌张力正常,腹壁和膝腱反射可引出,克氏征、布氏征、巴氏征均阴性。
血常规发现白细胞计数13.64×109/L(参考值:4~10×109/L),嗜中性粒细胞百分比35.6%(参考值:50%~70%),淋巴细胞百分比55.5%(参考值:20%~40%),血红蛋白107 g/L(参考值:110~170 g/L),血小板计数372×109/L [参考值:(100~200)×109/L]。生化检查总胆红素(Tbil)、结合胆红素(Dbil)和总胆汁酸(TBA)等胆汁淤积指标升高,但GGT 及胆固醇水平正常(表 1)。患儿及其父母行SLC25A13 基因高频突变筛查,未发现c.851del4、c.1638-1660dup、IVS6+5G>A、IVS16ins3kb 及IVS4ins6kb 等SLC25A13 基因高频突变。
1.2 全基因组测序(WGS)采集患儿静脉EDTA 抗凝外周血2 mL 按照blood DNA Mini kit 试剂盒(Simgen 公司,中国)说明书步骤提取基因组DNA 5 μg,并使用Covaris S2 超声仪将其打断扩增,纯化后得到富集的DNA文库。直接利用新一代测序仪IlluminaHiSeq2000(Illumina 公司,美国)进行测序,对测序产生的95.93G 原始数据进行去接头、过滤50% 的低质量序列处理,使Q20 值达93.33%,从而保证质量控制。最后进行基因序列生物信息学分析,找出致病基因及其突变性质。突变命名采用人类基因组变异协会HGVS(http://www.hgvs.org)命名法,变异意义参考Ensemble 数据库(http://www.ensembl.org)。
1.3 Sanger 测序验证采用套式PCR 扩增患儿及其父母ATP8B1基因的外显子18 及其侧翼序列,扩增及测序引物根据ATP8B1 基因的序列(Ensemble Genome Browser:ENSG00000283684) 运用Premier 5.0 软件设计,由上海立菲生物技术有限公司合成(表 2)。第1 次PCR 扩增的反应体系为50 μL,包含36.75 μL 灭菌双蒸水,5 μL 10×Buffer,4 μL dNTP,1 μL 引物ATPE×18F 和ATPE×18R,0.25 μL rTaq DNA 聚合酶(TakaRa),2 μL DNA;扩增条件为94℃预变性5 min,94℃变性30 s,46℃退火40 s,72℃延伸40 s,20 个循环,最后再72℃延伸10 min。第2 次PCR 扩增的反应体系为50 μL,包含了38.25 μL 灭菌双蒸水,5 μL 10×Buffer,4 μL dNTP,1 μL 引物ATPE×18F2 和ATPE×18R2,0.25 μL rTaq DNA 聚合酶(TakaRa),0.5 μL 第1 次PCR 产物;扩增条件为94℃预变性5 min,94℃变性30 s,46℃退火40 s,72℃延伸40 s,35个循环,最后再72℃延伸10 min。
| 表 2 ATP8B1 基因突变位点的PCR 扩增及Sanger 测序引物 |

PCR 产物经2% 琼脂糖凝胶电泳分离,目的条带使用切胶纯化试剂盒(Qiagen)从胶中回收。将纯化的PCR 产物送往英潍捷基(上海)贸易有限公司进行测序。应用Chromas 软件对测序结果进行分析,并应用DNAMAN 软件与ATP8B1基因组参考序列(Ensemble Genome Browser:ENST00000283684)进行比对。
本研究经暨南大学附属第一医院医学伦理委员会批准,并获得患儿父母知情同意。
2 结果 2.1 遗传学分析结果WGS 分析未发现PFIC2(ABCB11)、胆汁淤积综合征(VPS33B 和VIPAR)、家族性阿米什高胆烷血症(TJP2)、家族性高胆烷血症(EPHX1)、原发性胆汁酸合成或结合缺陷(HSD3B7、AKR1D1、CYP7B1、BAAT 和SLC27A5 等) 等以血清GGT 水平不高为生化特点的遗传性胆汁淤积症相关基因的任何致病突变,但在患儿ATP8B1基因外显子18 检测到1 个纯合突变c.2081T>A(p.I694N)(图 1)。该错义突变引起FIC1 蛋白第694 位氨基酸残基由异亮氨基酸变为天冬酰胺,为国外文献已报道的致病性突变[12-13],但尚未见于国内文献。Sanger 测序验证结果证实,患儿为突变c.2081T>A(p.I694N)的纯合子,而其父母均为该突变的携带者(图 2)。

|
图 1 ATP8B1 基因纯合突变c.2081T>A(p.I694N) 全基因组测序结果 红框内为ATP8B1 基因c.2081 位点WGS 分析结果,可见野生型胸腺嘧啶T 全部突变为腺嘌呤A。 |

|
图 2 患儿及其父母ATP8B1 基因Sanger 测序验证结果 A:患儿为c.2081T>A(p.I694N) 纯合突变;B:父亲为c.2081T>A 突变携带者;C:母亲为c.2081T>A 突变携带者。 |
2.2 治疗与结局
根据患儿临床症状体征及实验室检查结果,诊断考虑遗传性胆汁淤积症,但限于当时技术条件,未能对具体病因做深入研究。患儿1 岁8 个月时,在某医院临床诊断Byler 综合征,行胆囊结肠Roux-en-Y 吻合术,术后服用熊去氧胆酸和葡醛内酯等药物,皮肤巩膜黄染迅速消退。患儿5 岁11 个月时,经WGS 及Sanger 测序验证,最终确诊为ATP8B1 基因突变导致的PFIC1。电话随访至6 岁,黄疸未再出现反复,但肝功能和肝大改善情况不详。
3 讨论WGS 分析具有较好的均一性和覆盖度,能同时对20 000 种基因进行突变筛查,特别是对单核苷酸变异或小片段插入、缺失的检测具有明显优势 [14-15]。对于没有典型临床特征或确切候选基因的孟德尔遗传性病,WGS 可作为临床第一线测试手段[16]。然而,WGS 对多碱基重复、基因倒位重排,以及长片段的插入、缺失等突变类型的检出率较低,约为25%[17]。因此对于WGS 发现的突变,需采用一代测序技术进一步验证,以保证基因检测的准确性和有效性[18]。本例患儿临床表现并不十分典型,仅靠临床特点也难以确定致病基因,但借助WGS 技术,排除了PFIC2、胆汁淤积综合征、家族性阿米什高胆烷血症、家族性高胆烷血症、原发性胆汁酸合成或结合缺陷等具有类似临床和生化特征的遗传性胆汁淤积症,同时发现该患儿为ATP8B1 错义突变纯合子,经Sanger 测序验证结果与WGS 一致,确保了PFIC1 分子诊断结果的可靠性。值得注意的是,外显子捕获或者靶向Panel 高通量测序尽管测序覆盖范围低于WGS,但具有检测成本低和数据分析速度快等优势,在胆汁淤积症遗传学病因的识别方面可能具有良好的应用前景。
ATP8B1 编码的FIC1 蛋白的保守区包括驱动区、磷酸化区和核苷酸结合区,其中I694 位于核苷酸结合区的β 链下游,靠近驱动区,与FIC1 蛋白折叠相关。本例患儿ATP8B1 突变点为c.2081T>A,引起位于FIC1 蛋白胞质结构域的第694 位氨基酸残基由异亮氨基酸变为天冬酰胺(p.I694N),导致FIC1 蛋白异常折叠,影响FIC1蛋白的P 型ATP 酶活性[12-13, 19],从而导致患儿出现黄疸、皮肤瘙痒和肝脾肿大等肝内胆汁淤积表现,但其具体机制尚不完全清楚。有学者认为可能由于胆小管膜上磷脂的不对称性分布状态受损,增强了疏水性胆盐提取胆小管膜胆固醇的敏感性,影响到胆盐输出泵(BSEP)转运胆盐,从而出现胆汁淤积[2]。亦有学者认为,阴离子胆盐在胆小管的分泌形成了负电荷跨膜流动,而FIC1 可能作为一种阳离子挤压泵(cation extrusion pump),起到代偿上述负电荷流动的作用。因此,ATP8B1 突变造成FIC1 蛋白功能缺陷,导致肝细胞内离子失平衡,从而触发胆汁淤积[20]。此外,由于FIC1 蛋白在肠、肾和胰腺等组织上均有表达,故PFIC1 患儿多有腹泻、胰腺炎、神经性耳聋、身材矮小和佝偻病等肝外表现[2, 8],个别病例出现苔藓样硬化、手脚肿大的表现[21]。本例患儿肝内胆汁淤积特征明显,但未发现上述肝外症状体征。随着年龄增长,肝外表现是否会出现,有待随访观察。
本例患儿血清胆汁酸、直接胆红素、总胆红素、转氨酶等生化值均升高,符合肝内胆汁淤积症实验室特点。血清GGT 是重要的胆汁淤积指标,但此患儿多次血生化检查GGT 水平均未见升高,这有别于一般胆汁淤积症。PFIC1 患者血清GGT 不增高的原因尚不清楚,但有学者认为,生理状态下GGT 通过糖化磷脂酰肌醇(glycosyl phosphatidyl inositol)锚定于肝脏胆小管膜上,依赖管腔内高浓度胆汁酸盐的去污剂活性而释放到管腔;ATP8B1基因突变可导致BSEP 功能障碍,使胆汁酸分泌受损,而胆汁中低浓度的胆汁盐无法将锚定于胆小管膜上的GGT 洗脱下来并返流入血,因而PFIC1患儿血清GGT 不会增高[6]。
PFIC1 患儿的内科治疗主要采用熊去氧胆酸、利福平等药物,但具有一定不良反应且临床疗效不佳[22]。有报道认为肝移植为唯一可能治愈PFIC的治疗措施,但肝移植后PFIC1 患儿的肝外症状未见明显改善,部分患儿甚至进一步加重[7]。Diao等[23] 通过对20 例患者的临床研究证实了腹腔镜胆囊结肠吻合术治疗本病的可行性与安全性,认为胆囊结肠吻合术具有手术时间短、疗效显著以及术后并发症少等优点,是目前最有前景的治疗方法之一。本例患儿多次服用熊去氧胆酸和中药,效果均不明显,在行胆囊结肠Roux-en-Y 吻合术后,黄疸瘙痒等临床症状得以改善,也支持上述观点,但其远期结局仍需要长期临床观察。
综上所述,本例患儿通过WGS 及Sanger 测序验证,确诊为ATP8B1 基因突变导致的PFIC1。尽管患儿病程迁延,内科疗效欠佳,但经胆囊结肠Roux-en-Y 吻合术治疗,临床效果良好。此例PFIC1 患儿的诊治经过和遗传学特征研究,对于今后本病患儿的诊断治疗具有一定参考价值。
| [1] | Srivastava A. Progressive familial intrahepatic cholestasis type[J]. J Clin Exp Hepatol , 2014, 4 (1) : 25–36. DOI:10.1016/j.jceh.2013.10.005 |
| [2] | Paulusma CC, Oude Elferink RP, Jansen PL. Progressive familial intrahepatic cholestasis type 1[J]. Semin Liver Dis , 2010, 30 (2) : 117–124. DOI:10.1055/s-0030-1253221 |
| [3] | Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for:Progressive familial intrahepatic cholestasis type 1[J]. Eur J Hum Genet , 2014, 22 (4) . DOI:10.1038/ejhg.2013.186 |
| [4] | Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis[J]. Nat Genet , 1998, 18 (3) : 219–224. DOI:10.1038/ng0398-219 |
| [5] | van Mil SW, van Oort MM, van den Berg IE, et al. Fic1 is expressed at apical membranes of different epithelial cells in the digestive tract and is induced in the small intestine during postnatal development of mice[J]. Pediatr Res , 2004, 56 (6) : 981–987. DOI:10.1203/01.PDR.0000145564.06791.D1 |
| [6] | van Mil SW, Houwen RH, Klomp LW. Genetics of familial intrahepatic cholestasis syndromes[J]. J Med Genet , 2005, 42 (6) : 449–463. DOI:10.1136/jmg.2004.026187 |
| [7] | Lykavieris P, van Mil S, Cresteil D, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepaticFeatures:no catch-up of stature growth,exacerbation of diarrhea,and appearance of liver steatosis after liver transplantation[J]. J Hepatol , 2003, 39 (3) : 447–452. DOI:10.1016/S0168-8278(03)00286-1 |
| [8] | 徐志强, 张鸿, 董漪, 等. 进行性家族性肝内胆汁淤积症1型1例及文献复习[J]. 遗传病信息 , 2013, 26 (5) : 304–307. |
| [9] | 林瑞珠, 刘丽, 盛慧英, 等. 进行性家族性肝内胆汁淤积症1型1例[J]. 广东医学 , 2014, 35 (11) : 1807. |
| [10] | 刘圣烜, 黄志华, 董琛. 婴儿胆汁淤积症1106例临床分析[J]. 中国实用儿科杂志 , 2013, 28 (8) : 585–589. |
| [11] | 陆怡, 刘丽艳, 王晓红, 等. 低血清谷氨酸转肽酶进行性家族性肝内胆汁淤积症23例临床分析[J]. 中国循证儿科杂志 , 2012, 7 (8) : 1600–1604. |
| [12] | Chen HL, Chang PS, Hsu HC, et al. FIC1 and BSEP defects in Taiwanese patients with chronic intrahepatic cholestasis with low g-glutamyltranspeptidase levels[J]. J Pediatr , 2002, 140 (1) : 119–124. DOI:10.1067/mpd.2002.119993 |
| [13] | Liu LY, Wang XH, Wang ZL, et al. Characterization of ATP8B1 gene mutations and a hot-linked mutation found in Chinese children with progressive intrahepatic cholestasis and low GGT[J]. J Pediatr Gastroenterol Nutr , 2010, 50 (2) : 179–183. DOI:10.1097/MPG.0b013e3181c1b368 |
| [14] | Blue Cross and Blue Shield Association. Special report:exome sequencing for clinical diagnosis of patients with suspected genetic disorders[J]. Technol Eval Cent Assess Program Exec Summ , 2013, 28 (3) : 1–4. |
| [15] | BelkadiA, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants[J]. Proc Natl Scad Sci U S A , 2015, 112 (17) : 5473–5478. DOI:10.1073/pnas.1418631112 |
| [16] | Rosenblatt DS. Who's on first in exome and whole genome sequencing? Is it the patient or the incidental findings?[J]. Mol Genet Metab , 2013, 110 (1-2) : 1–2. DOI:10.1016/j.ymgme.2013.06.001 |
| [17] | Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders[J]. N Engl J Med , 2013, 369 (16) : 1502–1511. DOI:10.1056/NEJMoa1306555 |
| [18] | Shigemizu D, Fujimoto A, Akiyama S, et al. A practical method to detect SNVs and indels from whole genome and exome sequencing data[J]. Sci Rep , 2013, 3 : 2161. |
| [19] | van der Velden LM, Stapelbroek JK, Krieger E, et al. Folding defects in P-type ATP8B1 associated with hereditary cholestasis are ameliorated by 4-phenylbutyrate[J]. Hepatology , 2010, 51 (1) : 286–296. DOI:10.1002/hep.23268 |
| [20] | van Mil SW, Klomp LW, Bull LN, et al. FIC1 disease:a spectrum of intrahepatic cholestatic disorders[J]. Semin Liver Dis , 2001, 21 (4) : 535–544. DOI:10.1055/s-2001-19034 |
| [21] | Doğancı T, Akyol G, Bulac S. Progressive familial intrahepatic cholestasis with normal GGT level appearing with lichenification and enlargement of hands and feet[J]. Turk J Pediatr , 2005, 47 (4) : 385–389. |
| [22] | Jacquemin E, Hermans D, Myara A, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis[J]. Hepatology , 1997, 25 (3) : 519–523. DOI:10.1002/(ISSN)1527-3350 |
| [23] | Diao M, Li L, Zhang JS, et al. Laparoscopic cholecystoco-lostomy:a novel surgical approach for the treatment of progressive familial intrahepatic cholestasis[J]. Ann Surg , 2012, 258 (6) : 1028–1033. |