 2016, Vol. 18
2016, Vol. 18



微缺失和微重复综合征(microdeletion and microduplication syndromes, MMSs)是基因组重排导致的一类疾病,由特定染色体区域的拷贝数变异(copy number variations, CNVs)引起,包括DNA片段的缺失和重复,从而引起亚显微水平的基因量变化,DNA片段大小可以从几个kb到数个Mb。在2000年以前,只有几十种MMSs被发现,随着全基因高分辨分析技术的发展,特别是染色体微阵列(chromosomal microarray analysis, CMA)又称为染色体芯片在人类基因研究和诊断领域的广泛应用,目前全球已报道了近300种MMSs[1]。研究发现,MMSs可以导致全身多系统功能障碍,与精神发育迟滞(mental retardation, MR)、发育迟缓(developmental delay, DD)、多发畸形及自闭症等[2-3]。本研究应用CMA技术诊断MMSs 50例,并分析该50例患儿的临床表现及致病CNVs的特点。
1 资料与方法 1.1 研究对象回顾分析2013年6月至2015年9月我科儿童神经及内分泌专科诊断的50例MMSs患儿,通过CMA均诊断为致病性CNVs。所有患儿均完成血常规、尿常规、肝肾功能、甲状腺功能、生长激素、血氨基酸和酰基肉碱谱分析、尿有机酸分析等检测,结果均在正常范围,排除了常见内分泌及遗传代谢性疾病。
本研究获得医院伦理委员会批准。所有患儿监护人均签署知情同意书。
1.2 研究方法根据不同的年龄段,分别采用《Gesell发育量表》和《中国韦氏智力量表》测定发育商(DQ)或智商(IQ),同时运用《婴儿-初中生社会适应性能力量表》进行社会适应性能力评价。IQ或DQ < 70,伴社会适应性能力低下者诊断为MR或DD,≥5岁者称为MR, < 5岁者称为DD,以下统称为MR/DD。运用视觉诱发电位及听性脑干反应评价其视力和听力情况。
1.3 检测方法应用Affymetrix公司的CytoScan HD全基因芯片扫描技术检测全基因组CNVs。按试剂盒说明书操作,将样本依次进行PCR扩增、提纯、定量,片段化处理,再将DNA溶液标记后加入杂交试剂孵育,载入到芯片中进行杂交反应;洗染芯片后扫描分析。用Affymetrix GeneChip Scaner生成原始cel文件,通过AGCC软件芯片图像显示分析,利用CHAS软件分析结果。针对所发现的罕见CNVs,尽可能采用双亲样本重复CMA检测,了解其是否为新生CNV。
1.4 CNVs评估方法对于检测出的CNVs,首先查询UCSC、DECIPHER、ISCA、PubMed、CNVs多态性数据库(database of genomic variants)、既往文献及在线人类孟德尔遗传数据库(online Mendelian inheritance in man, OMIM),均确认为致病性CNVs。
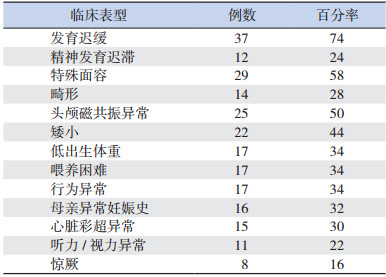
2 结果 2.1 研究对象的临床表型特点50例MMSs患儿中男性30例、女性20例,男女比例为1.5:1,年龄1个月至19岁。临床表型主要包括MR/DD、矮小、特殊面容/骨骼畸形、行为异常,同时合并母亲异常妊娠史以及低出生体重、喂养困难、惊厥发作及多系统异常等。患儿临床表型常涉及多个系统,各类临床表型所占比例亦有所差别,其中MR/DD、特殊面容/骨骼畸形、头颅磁共振异常及矮小是最常出现的临床表型,而惊厥、视力及听力的损害发生几率相对较小。见表 1。
| 表 1 50例MMSs的临床表型分类及所占比例 |
2.2 CNVs的结果及数据比对分析
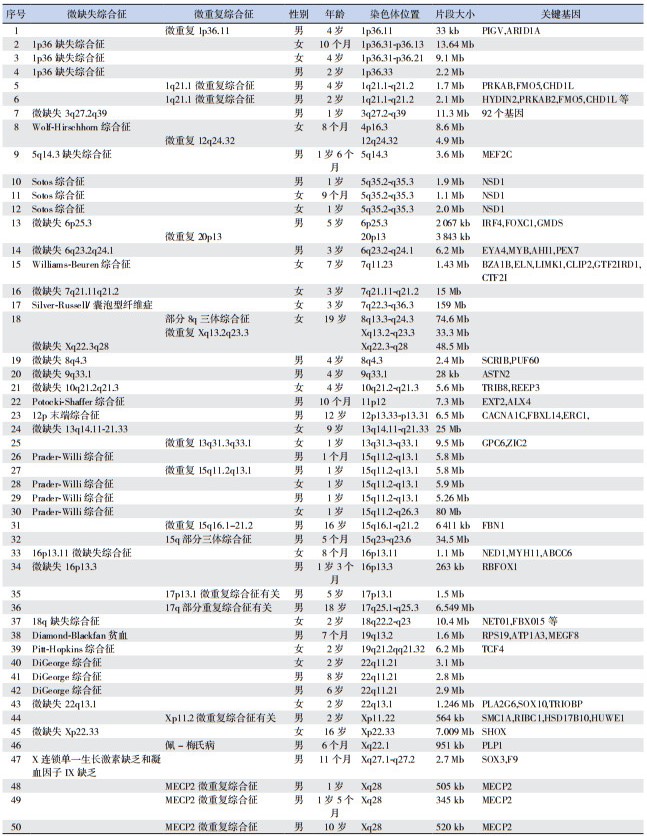
50例MMSs患儿共存在54处致病性CNVs,微缺失片段36个,微重复片段18个,比例为2:1,片段大小介于28 kb至48.5 Mb,平均13.86 Mb。发生在1号染色体的致病性CNVs共6处,其中3例1p36微缺失综合征;5号染色体上致病性CNVs共4处,其中3例为Sotos综合征;15号染色体上致病性CNVs共7处,其中4例为Prader-Willi综合征;22号染色体上致病性CNVs共5处,其中3例为DiGeoge综合征;X染色体上的致病性CNVs共8例9处,其中3例为MECP2微重复综合征。见表 2。
| 表 2 50例MMSs致病CNVs结果分析 |
3 讨论
本研究50例MMSs患者中,常见表现以MR/DD、矮小、特殊面容为主,且往往多系统的异常同时存在,且发现50%MMSs患儿存在头颅磁共振异常,包括侧脑室稍增宽、胼胝体发育不良、髓鞘化不良等,可能与研究对象年龄尚小,神经系统发育不完善有关,此点既往文献未见报道,仍需进一步大样本研究分析。目前MR/DD仍是严重的社会问题,发病率在2%~3%,50%的患者无法明确病因[4]。近年来研究发现,MR/DD患儿,尤其是伴随其他异常症状,如特殊面容、先天畸形、自闭症等,可能存在致病性的CNVs[5-6],国外报道不明原因的MR/DD中致病性的CNVs检出率为5.6%~20%[7]。余永国[8]对500例不明原因MR/DD患儿行CMA检测,发现158例(31.6%)存在致病CNV;我中心曾在55例不明原因的MR/DD患儿中,发现21例为MMSs,阳性率为38.2%[9]。因此MMSs为不明原因的MR/DD提供了重要的病因诊断,但MMSs的临床症状缺乏特异性,临床特点包括MR/DD、矮小、特殊面容、多发畸形、自闭症谱系障碍以及母亲异常妊娠史等[2-3],且与CNVs的片段大小和外显率有关,因此难以通过临床症状区分MMSs。比如本研究中3例1p36微缺失综合征,根据缺失片段大小,临床表型不尽相同,缺失片段越大,临床表型可能越多。有学者认为,对于具有相似或重叠临床表型的患者,可以采取基因型优先的方法来明确诊断,这种方法相对于以往表型优先的方法,能更有效快速地发现致病的细胞遗传学异常,为临床医生及时发现其他相应的临床表型提供指引[10]。
在减数分裂中,染色体发生不平衡交换,形成携带有CNV的配子,并进一步传递给后代,因此在理论上,每一种微缺失综合征都应该有与之相反的或者镜面关系的微重复综合征。然而,目前全球报道了211种微缺失综合征,仅有79种微重复综合征,在267个不同的基因位点中的比例为2.5:1,其中只有56个基因位点(21%)存在互为镜面的关系的MMSs [1]。本研究50 MMSs中36种微缺失,而只有18种微重复,比例为2:1,略低于文献报道的比例,可能与样本量较少有关。导致这一微缺失和微重复数量差别的原因有很多,首先相对于微缺失,与之呈镜面关系的微重复出现的临床症状可能比较轻微,甚至完全不出现临床表型,因此临床表现不明显,就诊率低。事实上,就染色体水平而言,染色体三倍体或者多倍体较单倍体更容易存活[11]。其次,在减数分裂时,早期配子也会更加倾向发生缺失或者重复中的一个[12]。此外,根据Weise等[1]总结的目前已知的MMSs中,致病性CNVs多位于X染色体、2号染色体、15号染色体、17号染色体或16号染色体,本研究提示X染色体、15号染色体也容易出现致病性CNVs,可能由于X连锁多影响智能发育,以及与不同染色体稳定性有关。
在减数分裂期间,染色体内部或者染色体之间发生重排的基础是人类基因组中含有不计其数的重复序列,这种不稳定性不仅可以导致CNVs,同时又可以获得新的基因功能,这在人类基因进化或多样性方面起着重要的作用[13]。但是对于基因组上的重复序列来说,除了同一位点上的同源序列以外,不同位点的重复序列也可能具有高度的同源性,从而发生非等位基因同源重组(non-allelic homologue recombination, NAHR)。NHAR是突变率较高的CNVs发生的主要机理,是由两条同源的、但在基因组不同位置重复出现的、高度相似性的DNA序列配对并发生序列交换造成的。相同染色体上的同向重复序列间的NAHR会导致重复或缺失,反向重复序列间NAHR会导致倒位。其次,突变率低的CNV断裂点常位于重复序列内或者周围,多源自非同源末端接合(non-homologous end joining, NHEJ),NHEJ是一种双链DNA断裂后的修复机制,不受特殊序列的直接调控,也不需要具有同源性的DNA片段作为重组底物。此机制的修复蛋白可以直接将双股裂断的末端彼此拉近,再藉由DNA连接酶的帮助,将断裂的两股重新接合,这些断裂点正是MMSs发生的位点[14]。还有诸如DNA复制叉停滞和模板转换机制,该基因组重排机制是由于细胞分裂的过程中,DNA复制突然暂停,但是却没有在停下的地方重新开始,而是转换至另一个模板开始复制。通常这个现象发生在基因组中的一些特殊结构处,这些结构是由很多核苷酸重复序列构成的,如回文结构和十字DNA结构。而事实上,正是这一特殊结构可以协助模板链的转换[15]。已有的研究发现在佩梅病[15]、Smith-Magenis综合征或Potocki-Lupski综合征[16]、MECP2微重复综合征[17]等位点上都存在由于DNA复制错误所导致的CNVs,该机制可以解释具有复杂结构的CNVs,有可能成为发现罕见MMSs的突破点。
CNVs具有良性、致病性和未知临床意义基因组结构变异。多重对照研究显示每个人都携带许多拷贝数变异,大部分是良性的。如果父母的CNVs是良性的,是否在遗传过程中诱发子代发生致病性的CNVs,往往需要进一步证实。很多标准有利于解读CNVs的临床意义,包括遗传方式、重组类型(缺失或者重复)、片段大小以及所包含的功能基因[18]。新生突变(de novo)的CNVs较遗传的CNVs更有可能是致病性的,尤其是伴有严重异常的病例。当然也有一些例外情况,如1q21.1综合征,即使是遗传与表型正常的父母,也可能会导致严重的精神发育异常[19]。由于经济及其他社会原因,很多时候无法检测到父母的CNVs,一般来说,大片段的CNVs较小片段的CNVs更容易致病,因为较大片段的CNVs可能包含更多的基因,从而使剂量敏感元件发生改变的几率增加。缺失所致的基因剂量不足更易导致基因功能的缺失,而重复的解读较缺失更加复杂。包含大量功能基因或者已知疾病基因的CNVs相对于包含少量基因或者意义不明确基因的CNVs更容易被确定为致病性的。
本研究发现罕见de novo 15q15.1-21.2微重复,其父母均为正常CNVs,主要表现为严重的身材矮小、精神发育迟滞、语言发育迟缓,阴茎短小,前额增宽、低耳位、眼距增宽、人中短、嘴唇肥厚等特殊面容。该患儿在染色体15q15.1-21.2发生了6 411 kb的重复,包含了关键基因:FBN1、EID1、SEMA6D等。据文献报道,不同的FBN1基因突变类型导致不同临床表型,但均与骨骼病变相关,可以解释该患儿严重的身材矮小、骨龄延迟以及面容异常[20-21];EID1基因在神经系统中过表达可能会损伤认知及记忆功能[22],同时与SF-1基因有协同作用,SF-1在肾上腺及性腺发育中起着重要的作用;SEMA6D基因的过表达也会影响神经系统的发育,与其在维持神经系统稳定性及损伤修复中的作用有关[23]。
随着基因组检测技术的快速发展,越来越多人类基因组中的CNVs将被大量发现,以往认为是良性或是临床意义不明确的CNVs可能对某些复杂性疾病的致病具有潜在作用,而对于罕见的MMSs的识别及诊断更加具有挑战性,需要临床医生和细胞遗传学专家的进一步证实。
| [1] | Weise A, Mrasek K, Klein E, et al. Microdeletion and microduplication syndromes[J]. J Histochem Cytochem , 2012, 60 (5) : 346–358. DOI:10.1369/0022155412440001 |
| [2] | Fan YS, Ouyang X, Peng J, et al. Frequent detection of parental consanguinity in children with developmental disorders by a combined CGH and SNP microarray[J]. Mol Cytogenet , 2013, 6 (1) : 38. DOI:10.1186/1755-8166-6-38 |
| [3] | Kaminsky EB, Kaul V, Paschall J, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities[J]. Genet Med , 2011, 13 (9) : 777–784. DOI:10.1097/GIM.0b013e31822c79f9 |
| [4] | Coulter ME, Miller DT, Harris DJ, et al. Chromosomal microarray testing influences medical management[J]. Genet Med , 2011, 13 (9) : 770–776. DOI:10.1097/GIM.0b013e31821dd54a |
| [5] | Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay[J]. Nat Genet , 2011, 43 (9) : 838–846. DOI:10.1038/ng.909 |
| [6] | Battaglia A, Doccini V, Bernardini L, et al. Confirmation of chromosomal microarray as a first-tier clinical diagnostic test for individuals with developmental delay, intellectual disability, autism spectrum disorders and dysmorphic features[J]. Eur J Paediatr Neurol , 2013, 17 (6) : 589–599. DOI:10.1016/j.ejpn.2013.04.010 |
| [7] | Miller DT, Adam MP, Aradhya S, et al. Consensus statement:chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies[J]. Am J Hum Genet , 2010, 86 (5) : 749–764. DOI:10.1016/j.ajhg.2010.04.006 |
| [8] | 余永国. 染色体基因芯片分析技术在不明原因智力落后诊断中的应用——附500例基因芯片结果分析[J]. 诊断学理论与实践 , 2013, 12 (4) : 387–389. |
| [9] | 张丽娜, 梁立阳, 孟哲, 等. 应用微阵列比较基因组杂交技术对55例智力低下/发育迟缓患儿基因组拷贝数变异的分析[J]. 中国儿童保健杂志 , 2014, 22 (8) : 795–798. |
| [10] | Nevado J, Mergener R, Palomares-Bralo M, et al. New microdeletion and microduplication syndromes:A comprehensive review[J]. Genet Mol Biol , 2014, 37 (1 Suppl) : 210–219. |
| [11] | Liehr T, Ewers E, Hamid AB, et al. Small supernumerary marker chromosomes and uniparental disomy have a story to tell[J]. J Histochem Cytochem , 2011, 59 (9) : 842–848. DOI:10.1369/0022155411412780 |
| [12] | Turner DJ, Miretti M, Rajan D, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders[J]. Nat Genet , 2008, 40 (1) : 90–95. DOI:10.1038/ng.2007.40 |
| [13] | Gazave E, Darré F, Morcillo-Suarez C, et al. Copy number variation analysis in the great apes reveals species-specific patterns of structural variation[J]. Genome Res , 2011, 21 (10) : 1626–1639. DOI:10.1101/gr.117242.110 |
| [14] | Mrasek K, Schoder C, Teichmann AC, et al. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones[J]. Int J Oncol , 2010, 36 (4) : 929–940. |
| [15] | Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders[J]. Cell , 2007, 131 (7) : 1235–1247. DOI:10.1016/j.cell.2007.11.037 |
| [16] | Vissers LE, Stankiewicz P, Yatsenko SA, et al. Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies[J]. Hum Genet , 2007, 121 (6) : 697–709. DOI:10.1007/s00439-007-0359-6 |
| [17] | Marijke Bauters, Hide Van Esch, Michael J. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair[J]. Genome Res , 2008, 18 (6) : 847–858. DOI:10.1101/gr.075903.107 |
| [18] | Miller DT, Adam MP, Aradhya S, et al. Consensus statement:chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies[J]. Am J Hum Genet , 2010, 86 (5) : 749–764. DOI:10.1016/j.ajhg.2010.04.006 |
| [19] | Brunetti-Pierri N, Berg JS, Scaglia F, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities[J]. Nat Genet , 2008, 40 (12) : 1466–1471. DOI:10.1038/ng.279 |
| [20] | Tsilou E, MacDonald IM. Weill-Marchesani syndrome. GeneReviews[M/OL]. Seattle (WA):University of Washington, Seattle.[2016-04-12]. http://www.ncbi.nlm.nih.gov/books/NBK1114. |
| [21] | Radonic T. Marfan syndrome[J]. Ned Tijdschr Tandheelkd , 2013, 120 (12) : 665–668. DOI:10.5177/ntvt.2013. |
| [22] | Liu R, Lei JX, Luo C, et al. Increased EID1 nuclear translocation impairs synaptic plasticity and memory function associated with pathogenesis of Alzheimer's disease[J]. Neurobiol Dis , 2012, 45 (3) : 902–912. DOI:10.1016/j.nbd.2011.12.007 |
| [23] | Park YY, Park KC, Shong M, et al. EID-1 interacts with orphan nuclear receptor SF-1 and represses its transactivation[J]. Mol Cells , 2007, 24 (3) : 372–377. |