 2017, Vol. 19
2017, Vol. 19



抗胸腺细胞球蛋白(antithymocyte globulin,ATG)联合环孢素A(CsA)为主的免疫抑制治疗(immunosuppressive therapy,IST)为目前儿童无同胞白细胞抗原(HLA)相合供者来源的重型/超重型再生障碍性贫血(severe/very severe aplastic anemia,SAA/VSAA)患者的一线治疗方案[1]。随着IST疗效的不断提高,包括阵发性睡眠性血红蛋白尿症(paroxysmal nocturnal hemoglobinuria,PNH)、骨髓增生异常综合征/急性髓系白血病(myelodysplastic syndrome/acute myeloid leukemia,MDS/AML)以及细胞遗传学改变在内的克隆演变等远期并发症逐渐被重视[2-3]。研究表明,SAA/VSAA患儿接受IST后恶性克隆演变的发生率远高于接受异基因造血干细胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT)治疗者[4]。发生包括MDS/AML及克隆性细胞遗传学异常等恶性克隆演变的AA患儿预后不良[5-7]。初诊时疾病的严重程度、接受重组人粒细胞集落刺激因子(G-CSF)等治疗以及ATG被认为是导致SAA/VSAA患者远期克隆演变的危险因素[4, 8]。现对我中心231例接受IST治疗的SAA/VSAA患儿的临床特征及治疗转归进行回顾性分析,进一步探索SAA/VSAA患儿IST远期克隆演变的发生率以及相关危险因素。
1 资料与方法 1.1 研究对象本研究纳入2000年2月至2014年5月中国医学科学院血液病医院儿童血液病诊疗中心接受IST治疗(ATG+CsA方案)的231例、初诊年龄小于18岁的SAA/VSAA患者,其中男性109例,女性122例,中位年龄6岁(9个月至18岁),所有患儿均因无同胞HLA全相合供者而未能接受一线allo-HSCT。
AA诊断参考国际再生障碍性贫血协作组诊疗意见[9];疾病的严重程度参考Camitta标准[10]。结合疾病病程及血液病诊断及疗效标准[11],我中心定义缓慢进展为SAA/VSAA(病程至少6个月)的患儿为SAA-II型,其余诊断为SAA-I型。所有患者常规行彗星及丝裂霉素实验以排除先天性骨髓衰竭,骨髓细胞形态学检查及细胞遗传学检查排除MDS,流式细胞术检测CD55/CD59表达及酸溶血试验以进行PNH鉴别。
1.2 治疗方案IST方案:(1)所有患者均接受兔源抗胸腺细胞球蛋白(rATG,Genzyme)静脉滴注,每日2.5~5.4 mg/kg,第1~5天。同时进行血清病反应预防:泼尼松每日1 mg/kg口服,第1~15天(第1~5天静脉输注等效剂量的地塞米松或氢化可的松),如无严重血清病反应,第16天开始逐渐减量,至第30天停用。(2)CsA每日3~5 mg/kg口服,并根据血药浓度、血清肌酐及胆红素水平调整剂量,至少6个月;对于获得治疗反应的患者根据血象每3个月进行CsA逐渐减量。(3)自第8天起皮下注射G-CSF,每次5 μg/kg,每周3次,至中性粒细胞绝对计数(absolute neutrophil count,ANC)维持在0.5×109/L以上,至少3个月。(4)149例患者接受雄激素口服治疗,其中6例患者口服达那唑,每日10 mg/kg;118例患者口服司坦唑醇,每日0.2 mg/kg;25例口服十一酸睾酮,每日4.0 mg/kg。根据血象调整用药。(5)支持治疗:输注红细胞及血小板以维持Hb≥60 g/L及PLT≥10×109/L。对于ANC≤0.5×109/L的患者予磺胺甲恶唑口服,对于有感染证据的粒细胞缺乏患儿根据病原学证据给予针对性抗感染治疗。所有患儿在粒细胞缺乏期间均于千级层流病房内接受治疗。
1.3 疗效标准参考文献标准[11-12]评价疗效,定义完全缓解(CR)和部分缓解(PR)为获得治疗反应,未缓解(NR)为未获得治疗反应。IST后6个月内死亡的患者被定义为早期死亡。各时间点总体治疗反应率为每个随访时间点获得治疗反应的病例数占总体随访到可参与疗效评患儿例数的百分比。
1.4 随访231例初诊SAA/VSAA患者,随访截至2016年4月1日或患者死亡,中位随访时间为69(1~192)个月,14例患者(6.1%)失访。217例患者有完整的随访资料,包括血常规、骨髓细胞形态学及相关检查、肝肾功能、溶血相关检查、流式细胞术糖基化磷脂酰肌醇锚连接蛋白(GPI-AP)缺失比例测定、感染及血制品输注情况。总体生存期(overall survival,OS)定义为初诊至随访截止日期;无治疗失败生存期(failure-free survival,FFS)定义为初诊至发生以下任意一件被认为治疗失败事件的时间:克隆演变,失去已获得的治疗反应,以及其他原因导致的死亡。
克隆演变的判断参考2008年WHO诊断分型标准[13],包括治疗、随访过程中发生的染色体异常(至少2个中期分裂象)、PNH(流式GPI-AP缺失比例20%以上,伴血管内溶血的临床或实验室证据)以及MDS/AML转化。克隆演变的累积发生率曲线为与非进展相关死亡率竞争性分析的结果。
1.5 统计学分析采用SPSS 23.0软件及R软件3.2.1进行统计学处理。非正态分布的计量资料采用中位数(范围)表示,计数资料采用百分率表示。率的组间比较采用卡方检验,生存分析采用Kaplan-Meier法及Log-rank检验,并将单因素分析结果中P<0.1的变量纳入多因素分析,多因素分析采用Cox回归模型。P≤0.05为差异有统计学意义。
2 结果 2.1 临床特征231例患儿的中位ANC为 0.26(0.00~0.78)×109/L,中位网织红细胞绝对计数(absolute reticulocyte count,ARC)9.40(0.0~70.1)×109/L,中位PLT 9(0~56)×109/L;231例中SAA 135例、VSAA 96例,其中19例为SAA-II型。初诊时14例(7.4%)患者检测到至少2个中期分裂象的染色体异常,95例患者外周血粒细胞或红细胞可检测到PNH克隆,外周血淋巴细胞亚群检测提示49例(24.6%)患儿CD3+T细胞/淋巴细胞比值>80%。除SAA-Ⅱ型患者以外,其他患儿在接受IST前均未接受过治疗;所有患者发病至开始IST的中位时间为47(10~375)d,接受rATG的中位剂量为每日3.4(2.4~5.4)mg/kg。
2.2 血液学缓解与总体生存情况231例初诊年龄小于18岁的SAA/VSAA患者,截至末次随访,死亡42例(其中18例为早期死亡)。早期死亡患儿不参与治疗6个月后的疗效评定,治疗后3、6、9、12个月总体治疗反应率分别为18.6%(43/231)、34.3%(79/231)、51.6% (110/213)、60.6%(129/213)。Kaplan-Meier分析提示患儿5年OS率为82.7%(95%CI: 77.6 %~ 87.8%),5年FFS率为59.5%(95%CI: 53.0% ~ 66.0%)。在213例可评价疗效的患儿中,155例(72.8%)患儿获得IST治疗反应,153例至末次随访存活;58例(27.2%)患儿未获得治疗反应。58例IST无效患儿中27例(27/58)接受了allo-HSCT,其中25例(25/27,92.6%)成功植活并长期生存;余31例(31/58)未获得治疗反应的患儿接受包括口服CsA、雄激素及血制品输注等姑息性治疗,仅11例(11/31,35.5%)患儿至末次随访存活。
2.3 IST后克隆演变至末次随访,155例IST有效患儿中8例(5.2%)发生克隆演变,58例IST无效患儿中7例(12.1%)发生克隆演变,总体克隆演变累积发生率为6.5%(15/231),发生克隆演变的中位时间为IST后33(6~74)个月。
15例发生克隆演变的患儿中AML克隆演变5例,其中1例接受allo-HSCT治疗,至末次随访无病生存;2例接受化疗,未缓解;2例放弃治疗后死亡。发生MDS克隆演变的3例,其中1例诊断为难治性血细胞减少(MDS-RCC),未予任何治疗,末次随访时中度贫血伴血小板少;另2例进展为难治性血细胞减少伴原始细胞增多(MDS-RAEB),均接受allo-HSCT,至末次随访无病生存。2例发生7号染色体缺失,其中1例进展为AML,化疗无缓解、死亡;另1例无症状存活。1例发生+8染色体核型异常,至末次随访无症状存活。4例发生PNH克隆演变的患儿中,2例在初诊时可检测到少量PNH克隆(3.72%和4.65%);截至末次随访,3例存活,但依赖输注红细胞及口服CsA、糖皮质激素等治疗;1例患儿表现为中度贫血,未予特殊处理,存活。
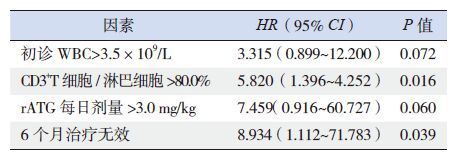
2.4 影响克隆演变的危险因素分析根据以往研究报道[5],将PNH及8号染色体三体核型异常(+8)定义为良性克隆演变,将MDS/AML及7号染色体异常缺失细胞遗传学改变(-7/7q-)定义为不良克隆演变。IST后良性克隆演变与不良克隆演变的5年累积发生率为(2.2±2.2)%与(4.8±3.3)%,见图 1。

|
图 1 231 例患儿IST 后克隆演变的累积发生率 |
为进一步探索儿童SAA/VSAA IST后不良克隆演变的危险因素,对患儿初诊临床资料及rATG剂量、6个月治疗反应与不良克隆演变发生率进行相关性分析。将初诊WBC>3.5×109/L、每日rATG剂量>3.0 mg/kg、初诊CD3+T细胞比例>80%以及与IST后6个月的治疗反应4个因素(表 1)纳入Cox多因素回归分析,提示初诊CD3+T细胞比例>80%、IST后6个月治疗无反应为IST后不良克隆演变的不利因素(表 2)。初诊CD3+T细胞比例>80%的患儿不良克隆演变的累积发生率高于CD3+T细胞比例≤80%者,差异无统计学意义(P=0.069),两者OS率的差异无统计学意义(P=0.794),见图 2A、2B。IST 6个月无效组的不良克隆演变累积发生率高于治疗有效组,但差异无统计学意义(P=0.095);其5年OS率[(82.9±7.1)%]低于治疗有效组[(100±0)%](P<0.01),见图 2C、2D。
| 表 1 IST 后SAA/VSAA 患儿发生不良克隆演变危险因素的单因素分析 [例(%)] |
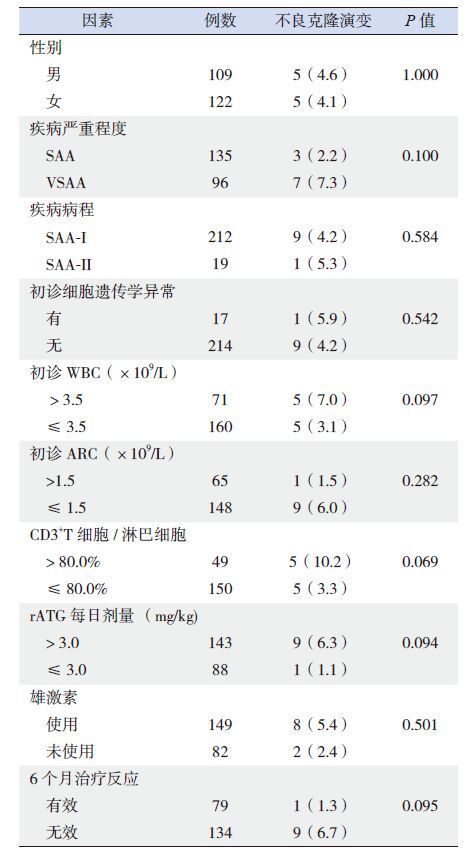
| 表 2 IST 后SAA/VSAA 患儿发生不良克隆演变危险因素的Cox 多因素分析 |

|
图 2 CD3+T 细胞比例与IST 后 6 个月治疗反应对不良克隆演变及总体生存的影响 |
3 讨论
AA是一类自身免疫介导的以骨髓造血功能衰竭、全血细胞减少为主要特征的获得性骨髓衰竭性疾病[14]。IST的疗效不断进步使越来越多SAA/VSAA患儿获得较长生存时间。但IST后AA患者的克隆演变成为临床医生逐渐认识并关注的重要并发症之一[2]。本研究6.5%的患儿发生IST后克隆演变,较既往研究发生率更低[15-17]。有研究认为GPI-AP缺失的细胞在体内并无生长优势,PNH克隆动态变化与AA患者的治疗反应及长期生存无关,也没有发现是否发生+8染色体核型异常的AA患者长期生存间的差异[5]。因此,我们将IST后PNH克隆演变及染色体+8定义为良性克隆演变。
随着近年来二代测序技术的不断发展,AA克隆性造血的研究不断深入,认为存在某些与髓系肿瘤相关体细胞基因突变的AA患者IST疗效较低[18-19],IST后治疗反应不佳的SAA/VSAA患者远期总体生存率也显著降低[20]。本研究显示,IST无反应的患者不良克隆演变发生率较高、OS率较低,IST后6个月无效为不良克隆演变的影响因素,与文献相符。由于本研究为回顾性研究,无法确认早期存在的克隆性体细胞突变是否与不良克隆演变相关,而克隆性造血的存在与IST后发生克隆演变的相关性仍不清楚,需要大规模临床实验进一步验证。
AA的发病机制主要与T淋巴细胞功能亢进介导的骨髓造血干/祖细胞的免疫损伤有关,因此导致造血细胞过度凋亡[21-22];而去除免疫抑制仍不能恢复骨髓造血集落的形成能力[23],提示免疫损伤后造血恢复功能可能受损。关于AA具体的免疫机制仍不十分清楚。有研究认为AA患者CD8+杀伤T细胞(CTL)水平在一定程度上反映了Fas抗原在AA中的表达情况及造血细胞凋亡情况[24],而CD4+Th1/Th2淋巴细胞比例的改变以及CTL异常激活可造成AA患者骨髓细胞过度凋亡[25]。本研究纳入的SAA/VSAA患儿中,初诊CD3+T细胞超过80%的患儿发生包括-7染色体异常、MDS/AML不良克隆演变的比例较高,提示异常的T淋巴细胞激活可能与抗原刺激后的基因突变及克隆性造血相关,然而造血干/祖细胞经历免疫损伤后是否发生克隆演变以及具体的病理生理机制仍然有待于进一步的实验室研究。
关于AA患者的G-CSF使用与IST后克隆演变发生的相关性还存在争议。Li 等[8]报道AA患者的G-CSF使用与恶性克隆演变相关;日本的研究也认为IST后发生MDS/AML克隆演变与G-CSF的使用有关[3];另有报道称G-CSF的使用与儿童AA远期发生染色体-7相关[7]。而Gurion等[26]通过对6项前瞻性研究进行Meta分析,证实G-CSF的使用与远期发生克隆演变无关。本研究仅2例患者发生IST后染色体-7的克隆演变,其中1例接受了G-CSF治疗。本研究结果还显示,G-CSF使用与否与AA的IST后克隆演变并无显著关联。但儿童克隆性造血的发生率低于成人患者[27-28],且本研究中患儿接受G-CSF的使用时间明显短于既往报道,故无法得出儿童AA患者G-CSF使用与远期克隆演变关系的结论。
一项来自欧洲骨髓移植协作组的回顾性研究报道,儿童SAA患者接受免疫抑制治疗后恶性克隆的发生率要显著高于接受亲缘供者来源的异基因造血干细胞移植SAA患儿[4]。表明长时间接受免疫抑制治疗本身可能是促使造血干细胞发生克隆性改变的原因。本研究虽然只纳入了接受了IST的患者,无法进行AA的两种一线治疗方案发生克隆演变的对照研究,但提示ATG每日剂量>3.0 mg/kg的患儿不良克隆演变的比例较高,尽管多因素分析无统计学意义,因而需要更大规模的前瞻性临床研究予以证实。
SAA/VSAA患儿IST后发生克隆演变作为被广泛认识到的远期并发症预后较差,尽早识别出发生克隆演变的高危患儿对于临床医生制定合理的诊疗决策至关重要。本研究中患儿初诊高比例的CD3+T细胞水平及免疫抑制治疗早期无效与IST治疗后远期发生恶性克隆演变显著相关,警惕以上危险因素并尽早选择二线治疗方案,有助于节约治疗花费并提高患儿的远期生存。
| [1] | Scheinberg P, Young NS. How I treat acquired aplastic anemia[J]. Blood, 2012, 120 (6): 1185–1196. DOI:10.1182/blood-2011-12-274019 |
| [2] | Tichelli A, Gratwohl A, Würsch A, et al. Late haematological complications in severe aplastic anaemia[J]. Br J Haematol, 1988, 69 (3): 413–418. DOI:10.1111/j.1365-2141.1988.tb02382.x |
| [3] | Kojima S, Ohara A, Tsuchida M, et al. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children[J]. Blood, 2002, 100 (3): 786–790. DOI:10.1182/blood.V100.3.786 |
| [4] | Dufour C, Pillon M, Socié G, et al. Outcome of aplastic anaemia in children. A study by the severe aplastic anaemia and paediatric disease working parties of the European group blood and bone marrow transplant[J]. Br J Haematol, 2015, 169 (4): 565–573. DOI:10.1111/bjh.13297 |
| [5] | Maciejewski JP, Risitano A, Sloand EM, et al. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia[J]. Blood, 2002, 99 (9): 3129–3135. DOI:10.1182/blood.V99.9.3129 |
| [6] | Kaito K, Kobayashi M, Katayama T, et al. Long-term administration of G-CSF for aplastic anaemia is closely related to the early evolution of monosomy 7 MDS in adults[J]. Br J Haematol, 1998, 103 (2): 297–303. DOI:10.1046/j.1365-2141.1998.01014.x |
| [7] | Ohara A, Kojima S, Hamajima N, et al. Myelodysplastic syndrome and acute myelogenous leukemia as a late clonal complication in children with acquired aplastic anemia[J]. Blood, 1997, 90 (3): 1009–1013. |
| [8] | Li Y, Li X, Ge M, et al. Long-term follow-up of clonal evolutions in 802 aplastic anemia patients:a single-center experience[J]. Ann Hematol, 2011, 90 (5): 529–537. DOI:10.1007/s00277-010-1140-9 |
| [9] | Incidence of aplastic anemia:the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study[J]. Blood, 1987, 70 (6): 1718–1721. |
| [10] | Camitta BM, Thomas ED, Nathan DG, et al. A prospective study of androgens and bone marrow transplantation for treatment of severe aplastic anemia[J]. Blood, 1979, 53 (3): 504–514. |
| [11] | 杨崇礼, 邵宗鸿.再生障碍性贫血[M]//张之南, 沈悌. 血液病诊断及疗效标准. 第3版. 北京:科学出版社, 2007:20-23. |
| [12] | Camitta BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia:a prospective studyof the effect of early marrow transplantation on acute mortality[J]. Blood, 1976, 48 (1): 63–70. |
| [13] | Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia:rationale and important changes[J]. Blood, 2009, 114 (5): 937–951. DOI:10.1182/blood-2009-03-209262 |
| [14] | Young NS. Current concepts in the pathophysiology and treatment of aplastic anemia[J]. Hematology Am Soc Hematol Educ Program, 2013, 2013 (1): 76–81. |
| [15] | Bacigalupo A, Bruno B, Saracco P, et al. Antilymphocyte globulin, cyclosporine, prednisolone, and granulocyte colony-stimulating factor for severe aplastic anemia:an update of the GITMO/EBMT study on 100 patients. European Group for Blood and Marrow Transplantation (EBMT) working party on severe aplastic nemia and the Gruppo Italiano Trapianti di Midolio Osseo (GITMO)[J]. Blood, 2000, 95 (6): 1931–1934. |
| [16] | Teramura M, Kimura A, Iwase S, et al. Treatment of severe aplastic anemia with antithymocyte globulin and cyclosporin A with or without G-CSF in adults:a multicenter randomized study in Japan[J]. Blood, 2007, 110 (6): 1756–1761. DOI:10.1182/blood-2006-11-050526 |
| [17] | Fuhrer M, Burdach S, Ebell W, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST):the SAA 94 experience. German/Austrian Pediatric Aplastic Anemia Working Group[J]. KlinPadiatr, 1998, 210 (4): 173–179. |
| [18] | Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic nemia[J]. N Engl J Med, 2015, 373 (1): 35–47. DOI:10.1056/NEJMoa1414799 |
| [19] | Ogawa S. Clonal hematopoiesis in acquired aplastic anemia[J]. Blood, 2016, 128 (3): 334–347. |
| [20] | Jalaeikhoo H, Khajeh-Mehrizi A. Immunosuppressive therapy in patients with aplastic anemia:a single-center retrospective study[J]. PLoS One, 2015, 10 (5): e0126925. DOI:10.1371/journal.pone.0126925 |
| [21] | Maciejewski JP, Risitano A. Hematopoietic stem cells in aplastic anemia[J]. Arch Med Res, 2003, 34 (6): 520–527. DOI:10.1016/j.arcmed.2003.09.009 |
| [22] | Chen Y, Zou Z, Wu Z, et al. TNF-α-induced programmed cell death in the pathogenesis of acquired aplastic anemia[J]. Expert Rev Hematol, 2015, 8 (4): 515–526. DOI:10.1586/17474086.2015.1049593 |
| [23] | Bacigalupo A, Podestà M, Mingari MC, et al. Immune suppression of hematopoiesis in aplastic anemia:activity of T-gamma lymphocytes[J]. J Immunol, 1980, 125 (4): 1449–1453. |
| [24] | 刘春燕, 郑萌颖, 付蓉, 等. 重型再生障碍性贫血细胞毒性T细胞免疫攻击靶点的体外实验[J]. 中华医学杂志, 2016, 96 (22): 1728–1732. |
| [25] | Sheng W, Liu C, Fu R, et al. Abnormalities of quantities and functions of linker for activations of T cells in severe aplastic anemia[J]. Eur J Haematol, 2014, 93 (3): 214–223. DOI:10.1111/ejh.12327 |
| [26] | Gurion R, Gafter-Gvili A, Paul M, et al. Hematopoietic growth factors in aplastic anemia patients treated with immunosuppressive therapy-systematic review and meta-analysis[J]. Haematologica, 2009, 94 (5): 712–719. DOI:10.3324/haematol.2008.002170 |
| [27] | Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence[J]. N Engl J Med, 2014, 371 (26): 2477–2487. DOI:10.1056/NEJMoa1409405 |
| [28] | Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes[J]. N Engl J Med, 2014, 371 (26): 2488–2498. DOI:10.1056/NEJMoa1408617 |