 2017, Vol. 19
2017, Vol. 19

2. 广州市妇女儿童医疗中心 遗传与内分泌科, 广东 广州 510180

 , LI Bei, JIANG Xiang, CHEN Qian-Yu, JIA Xue-Fang, TANG Cheng-Fang, LIU Li
, LI Bei, JIANG Xiang, CHEN Qian-Yu, JIA Xue-Fang, TANG Cheng-Fang, LIU Li
先天性甲状腺功能减低症(congenital hypothyroidism,CH)是由于甲状腺的分化、迁移或发育过程异常,或者是由于参与甲状腺激素合成过程中的基因突变,造成甲状腺激素合成不足的一种最常见的先天性内分泌疾病。我国CH的发病率约为1 : 2 050[1],广州市2011~2012年CH的发病率为1 : 2 779[2]。近年来研究显示,甲状腺激素合成障碍所致CH的比例呈增高趋势[3-4],其中以双氧化酶2(dual oxidase 2,DUOX2)基因突变最常见[2, 5]。自2008年意大利学者Zamproni等[6]首次报道双氧化酶成熟因子2(dual oxidase maturation factor 2,DUOXA2)基因突变性CH以来,DUOXA2与CH的关系越来越受到关注。本研究在前期CH分子流行病学研究基础上[2],对DUOX2基因检测未发现异常的20例疑似甲状腺激素合成障碍性CH患儿进行了DUOXA2基因突变分析,探讨广州地区DUOXA2基因的突变特点、基因型与表型的关系。
1 资料与方法 1.1 研究对象广州市新生儿筛查中心按照“先天性甲状腺功能减低症诊疗共识”[1]对广州市2011年1月1日至2012年12月31日出生的433 578例新生儿进行了CH筛查,确诊CH 156例,其中疑似甲状腺激素合成障碍性CH 118例(复查时甲状腺超声提示甲状腺正常或肿大者),本研究在前期研究(96例疑似甲状腺激素合成障碍性CH患儿中60例存在DUOX2基因突变、2例确诊为TPO基因突变所致永久性CH)[2]基础上,对其余34例CH患儿中的20例(59%,20/34)进行DUOXA2基因突变分析,其中男13例、女7例,无血缘关系。纳入标准:(1)新生儿干血滴纸片检测促甲状腺激素(bsTSH)≥9 mIU/L;(2)1个月内复查血清促甲状腺激素(sTSH)>15.00 mIU/L(4~30 天龄参考值:0.47~10.00 mIU/L)、游离甲状腺素(FT4)<15.00 pmol/L(参考值:12.00~29.34 pmol/L),超声提示甲状腺正常或肿大(校正椭圆法甲状腺体积[7],新生儿甲状腺体积参考值:0.6±0.2 mL);(3)甲状腺球蛋白(TG)正常或增高(参考值:3.50~77.00 ng/mL);(4)甲状腺自身免疫抗体TGAb、TPOAb均为阴性。随机选取50例无血缘关系、广州市2011~2012年出生、CH筛查阴性的干血滤纸样本作为对照,用于新突变验证。本研究获得广州市妇女儿童医疗中心伦理委员会批准及患儿监护人知情同意。
1.2 基因组DNA提取采集外周静脉血2 mL或干血滤纸标本,采用厦门致善生物科技有限公司Lab-Aid核酸分离试剂盒提取基因组DNA。
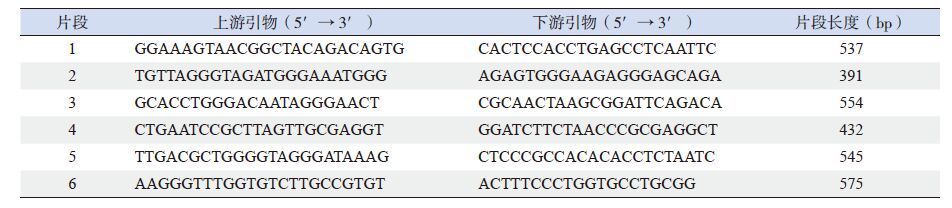
1.3 DUOXA2基因突变分析根据NCBI数据中DUOXA2基因系列(NM_207581.3),应用引物设计软件Primer 5.0设计6对引物(表 1),扩增DUOXA2基因所有外显子及其两侧侧翼序列,引物由北京六合华大基因股份有限公司合成。PCR反应体系:10×Buffer(Mg2+ plus)2.5 µL,dNTP Mixture(各10 mmol/L)2.0 µL,ExTaq聚合酶(2.5 IU/µL)0.125 µL,上下游引物(10 pmol/µL)各0.5 µL,模板DNA(80~100 ng)2.0 µL,加去离子水至25 µL。PCR反应条件:95 ℃预变性5 min,95 ℃变性30 s,64 ℃退火30 s,72 ℃延伸50 s,共35个循环;72 ℃再延伸10 min。扩增产物送至北京六合华大基因股份有限公司进行纯化、双向测序(ABl3730测序仪)。测序结果通过DNAMAN及Chromas软件与正常参考序列进行比对分析。发现突变则与人类基因突变数据库(HGMD)、千人基因组数据库、单核苷酸多态性数据库(dbSNP)以及近期文献进行突变位点的查找,明确是否为已报道突变或多态性;新突变在50例对照样本中进行检测,对新错义突变进行10个不同物种突变位点的氨基酸保守性分析,并进行SIFT和PolyPhen-2软件蛋白质功能预测。
| 表 1 DUOXA2 基因PCR 引物及扩增片段长度 |
1.4 再评估
所有CH患儿2~3岁时试停左旋甲状腺素1个月,复查甲状腺超声及血清TSH、FT4及TG水平。根据血清TSH及FT4水平判断临床转归:(1)暂时性CH,TSH及FT4正常;(2)永久性CH,TSH>15 mIU/L伴FT4 <15 pmol/L为典型的永久性CH,继续左旋甲状腺素替代治疗;如果TSH轻度增高(TSH 7~15 mIU/L)伴FT4正常,考虑为轻度的永久性CH,定期随访观察,必要时再次替代治疗。
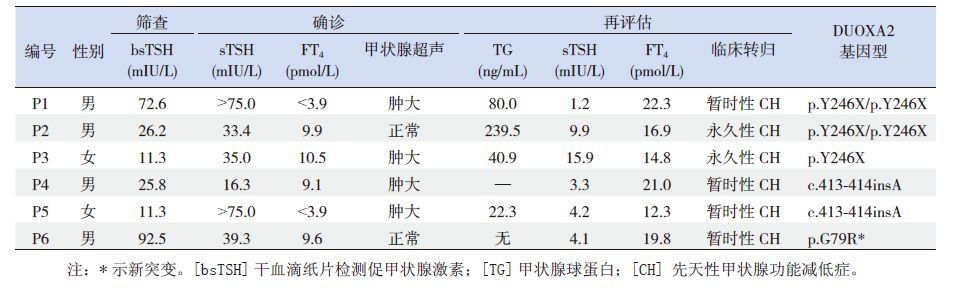
2 结果 2.1 DUOXA2基因分析在20例CH患儿中,6例检出DUOXA2基因突变,均无甲状腺相关疾病家族史,其中2例为已知突变:c.738C>G(p.Y246X)纯合突变;4例为携带者,分别为已知突变c.413-414insA(p.Y138X) 2例,p.Y246X及新突变c.235G>C(p.G79R)携带者各1例,见图 1。新突变p.G79R在50例对照组中未检出,10种不同物种氨基酸保守性分析提示新突变的氨基酸甘氨酸(G)高度保守(图 2),SIFT及PolyPhen2软件预测评分分别为0和0.997,均提示该突变影响蛋白质功能,致病可能性大。

|
图 1 6 例CH 患儿DUOXA2 基因突变的不同类型及正常对照测序图 ①、②分别为c.738C>G(p.Y246X) 纯合突变及正常对照;③、④分别为c.413-414insA(p.Y138X) 杂合突变及正常对照;⑤、⑥分别为新突变c.235G>C(p.G79R) 杂合突变及正常对照。箭头所指为突变位点。 |

|
图 2 新突变p.G79R 的氨基酸保守性分析 p.G79R的突变氨基酸(G)在10 种不同物种间高度保守。 |
2.2 DUOXA2基因型与表型的关系
6例DUOXA2基因突变患儿新生儿筛查时TSH>10 mIU/L,生后1个月内复查甲状腺功能提示血清FT4减低伴sTSH显著增高,超声检查显示4例伴有甲状腺肿大。2~3岁再评估显示:暂时性CH 4例,轻度及典型永久性CH各1例。
2例p.Y246X纯合突变患儿临床转归分别为暂时性CH(P1)和轻度永久性CH(P2),P2再评估后停药随访1年,sTSH在7~9.9 mIU/L、FT4正常,因TG显著增高,再次给予小剂量左旋甲状腺素替代治疗;4例携带者患儿中,除p.Y246X携带者(P3)表现为典型的永久性CH表型外,其余3例均为暂时性CH。见表 2。
| 表 2 6 例DUOXA2 基因突变的CH 患儿的基因型及临床表型特征 |
3 讨论
DUOXA2蛋白是内质网上的一种固有蛋白,对产生H2O2的关键酶DUOX2的转运、成熟及定位均发挥重要作用。DUOX2/DUOXA2系统中任何成员异常均会影响H2O2的合成,进而影响甲状腺过氧化物酶(TPO)的碘有机化过程,从而导致甲状腺激素合成障碍。
人类DUOXA2基因定位于染色体15q21.1,长约1.5 kb,含有6个外显子,编码320个氨基酸[8],呈常染色体隐性遗传。迄今为止,HGMD(http://www.hgmd.org)已收录了10种DUOXA2基因致病性突变,其中包括6种错义突变、1种无义突变、1种剪接突变、1种大片段缺失突变及1种插入突变。无义突变c.738C>G (p.Y246X)是中国人DUOXA2基因的最常见突变,该突变导致编码蛋白的第5跨膜螺旋和C端胞内结构域缺失,体外实验证实该突变导致DUOXA2蛋白功能的完全丧失[6]。Zamproni等[6]首次报道1名中国籍女性患者为p.Y246X纯合突变,临床表现为轻度的永久性CH,其携带杂合突变的父母及同胞表型正常。Liu等[9]报道了1例经新生儿筛查确诊的p.Y246X纯合突变所致的CH患者,伴甲状腺肿大,2岁试停左旋甲状腺素1个月再评估时表现为轻度永久性CH。本研究共发现3例患儿携带p.Y246X突变,其中2例p.Y246X纯合突变的患儿临床转归分别为暂时性CH和轻度永久性CH,而携带p.Y246X杂合突变的患儿表现为永久性CH。
c.413-414insA(p.Y138X)为插入移码突变,导致第138密码子TAC 转变为终止密码TAA,使DUOXA2蛋白缩短183个氨基酸。2013年我国学者Yi等[10]首先报道了该突变。随后,韩国学者Park等[5]对112例新生儿CH患者及58例门诊随访的CH患者进行了DUOX2与DUOXA2等基因的筛查,发现9例患者携带c.413-414insA突变,其中7例为c.413-414insA单等位基因杂合突变,2例为同时携带DUOXA2其他等位基因突变的复合杂合突变。目前尚未见c.413-414insA缺陷患者临床转归的文献资料。本研究显示,2例c.413-414insA携带者均为暂时性CH。
新突变c.235G>C(p.G79R)导致235位核苷酸发生G>C转换,使第79密码子由非极性甘氨酸变为极性精氨酸。该突变在HGMD数据库、千人基因组计划数据库、dbSNP数据库及正常对照组均未检出,其突变氨基酸甘氨酸(G)在10种不同物种中高度保守,SIFT及PolyPhen2软件预测,均提示该突变影响蛋白质功能,故p.G79R致病性可能性极大。
DUOXA2基因呈常染色体隐性遗传,但临床资料证实,DUOXA2单等位基因突变与CH发病有关[5, 9]。本研究进一步证实,DUOXA2基因双位点或单位点突变均可表现为暂时性CH或永久性CH,即使是相同的基因型(如p.Y246X纯合突变),其临床表型仍存在差异。DUOXA2基因突变的CH患者临床表型存在明显异质性的原因可能与以下几个因素有关:(1)体内存在着DUOXA1替代途径[8],DUOXA1表达约占DUOXA2的1/5,当DUOXA2蛋白活性丧失,DUOXA1蛋白能部分代偿[6];(2)环境因素,如碘缺乏或碘过多均可影响甲状腺激素的合成;(3)可能存在与CH相关的其他基因突变共存,例如与TPO、TG、TSHR等基因的突变共存。另外,由于一代测序方法的局限性,携带DUOXA2基因单等位基因突变的患儿可能存在发生在非编码区的另一等位基因的突变。
本研究在团队前期CH分子流行病学研究基础上[2],对DUOX2基因检测未发现异常的20例疑似甲状腺激素合成障碍性CH患儿进行了DUOXA2基因突变分析,6例检出DUOXA2基因突变。由此推测,广州地区DUOXA2基因突变所致新生儿CH的发病率约为1 : 36 131。
综上所述,DUOXA2基因突变在广州地区疑似甲状腺激素合成障碍性CH患儿中较常见,DUOXA2基因单位点突变或双位点突变均可导致暂时性CH及不同程度的永久性CH。新突变p.G79R为致病性突变的可能性大。明确CH病因,对指导治疗、预测疾病转归等可提供参考。
| [1] | 中华医学会儿科学分会内分泌遗传代谢学组, 中华预防医学会儿童保健分会新生儿疾病筛查学组. 先天性甲状腺功能减低症诊疗共识[J]. 中华儿科杂志, 2011, 49 (6): 421–424. |
| [2] | Tan M, Huang Y, Jiang X, et al. The Prevalence, clinical, and molecular characteristics of congenital hypothyroidism caused by DUOX2 mutations:A population-based cohort study in Guangzhou[J]. Horm Metab Res, 2016, 48 (9): 581–588. DOI:10.1055/s-0042-112224 |
| [3] | Hashemipour M, Ghasemi M, Hovsepian S, et al. Etiology of congenital hypothyroidism in Isfahan:Does it different[J]. Adv Biomed Res, 2014, 3 : 21. DOI:10.4103/2277-9175.124658 |
| [4] | Albert BB, Cutfield WS, Webster D, et al. Etiology of increasing incidence of congenital hypothyroidism in New Zealand from 1993-2010[J]. J Clin Endocrinol Metab, 2012, 97 (9): 3155–3160. DOI:10.1210/jc.2012-1562 |
| [5] | Park KJ, Park HK, Kim YJ, et al. DUOX2 mutations are frequently associated with congenital hypothyroidism in the Korean population[J]. Ann Lab Med, 2016, 36 (2): 145–153. DOI:10.3343/alm.2016.36.2.145 |
| [6] | Zamproni I, Grasberger H, Cortinovis F, et al. Biallelic inactivation of the dual oxidase maturation factor 2(DUOXA2) gene as a novel cause of congenital hypothyroidism[J]. J Clin Endocrinol Metab, 2008, 93 (2): 605–610. DOI:10.1210/jc.2007-2020 |
| [7] | 陈文敏, 邱文慧, 黄永兰, 等. 新生儿先天性甲状腺功能低下症的超声特征分析[J]. 临床超声医学杂志, 2016, 18 (7): 467–469. |
| [8] | Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent[J]. J Biol Chem, 2006, 281 (27): 18269–18272. DOI:10.1074/jbc.C600095200 |
| [9] | Liu S, Liu L, Niu X, et al. A novel missense mutation (I26M) in DUOXA2 causing congenital goiter hypothyroidism impairs NADPH oxidase activity but not protein expression[J]. J Clin Endocrinol Metab, 2015, 100 (4): 1225–1229. DOI:10.1210/jc.2014-3964 |
| [10] | Yi RH, Zhu WB, Yang LY, et al. A novel dual oxidase maturation factor 2 gene mutation for congenital hypothyroidism[J]. Int J Mol Med, 2013, 31 (2): 467–470. |