 2017, Vol. 19
2017, Vol. 19



2. 深圳市出生缺陷预防控制重点实验室, 广东 深圳 518000
结节性硬化症(tuberous sclerosis complex, TSC)属于常染色体显性遗传的神经皮肤综合征,发病率较低,约1/6 000~10 000,家族遗传约1/3[1]。以多组织、多器官的错构瘤损害为主要特征,常可累及大脑、皮肤、心脏、肾脏、肺和眼等[2]。临床表现各异,且具有年龄相关性,成人症状较儿童多,但除心脏错构瘤外[3]。其致病基因为TSC1和TSC2,二者均属肿瘤抑制基因,TSC1基因位于9q34,包含23个外显子,第1和第2外显子分别为非编码区和选择性剪接位点,编码产物为错构瘤蛋白(hamartin);TSC2基因位于16p13.3,包含41个外显子,编码产物为马铃薯蛋白(tuberin)。
TSC1和TSC2的致病突变包括无义突变、框移突变、剪接突变、缺失或插入和一些错义突变,二者均无突变热点。临床诊断为TSC的患者约85%经测序可检测到突变。测序阴性者,采用多重连接依赖性探针扩增技术(MLPA)检测大片段缺失和重排,TSC1和TSC2检出率分别为0.5%和6%[4]。本文就TSC2基因两个新发框移突变作如下讨论。
1 资料与方法 1.1 研究对象家系1先证者,女,4岁,无缺氧窒息史,母孕期无异常,1岁半独走。生后2个月于面部右侧颞部发现一色素脱失斑(图 1A)。生后6个月反复无诱因点头,4~6次/d,持续2~10 s/次。生后7个月颅脑MRI检查提示双侧侧脑室壁小结节灶及双侧大脑半球皮层下异常信号。1岁半复查CT提示双侧侧脑室壁边缘小结节灶较前有所增加(图 1B),双侧大脑半球皮层/皮层下小片状低密度灶,部分伴钙化,TSC可能。脑电图异常,清醒时双侧枕颞额大量尖慢波发放,临床痉挛发作伴高幅慢波出现。双肾B超未见异常。2岁时面部可见少量血管纤维瘤,逐渐增多。采集外周血送深圳华大基因研究院行TSC相关基因的高通量测序,结果提示TSC2基因c.3981-3982 insA(p.Asp1327AspfsX87)杂合突变。经解痉、抗癫癎治疗,现发作仍无减少,睡醒时易发作,有时玩耍中有张口、傻笑等动作,心跳加快,伴点头3~4次,发作后疲劳入睡,每天发作7~8次。有时继发双手僵硬,双眼斜视,喉中惊恐样发声,1~2次/周。现只会叫“爸爸”“妈妈”两个词且吐字不清,能听懂父母少部分话语,但不能执行指令,且有自闭症倾向。全身可见4处无规则色素脱失斑,服用雷帕霉素后,面部血管纤维瘤部分消退。先证者父母均无TSC相关症状。

|
图 1 家系1先证者皮肤改变和影像学特征 A:右侧颞部一片状色素脱失斑;B:1岁半头颅CT检查示右侧侧脑室旁2个点状高密度钙化灶(红色箭头所示),左侧侧脑室旁可见1个点状高密度钙化灶(黑色箭头所示)。 |
家系2先证者,女,9岁,出生史无异常,两颊面部见散在血管纤维瘤,腹部见钙化星形细胞错构瘤及小片状色素脱失斑。6岁时,间断无诱因抽搐2次,颅脑CT检查提示右侧侧脑室室管膜下见多发钙化灶影,考虑TSC。脑电图提示为界限,睡眠中见左侧中后颞区为主可疑尖波散发和睡眠背景欠佳。8岁时,颅脑CT检查提示两侧侧脑室旁多发钙化(图 2A),癫癎大发作共3次,双肾、心脏、肝、胆、胰、脾B超均未见异常。经抗癫癎治疗,效果较好。外周血经华大基因行高通量测序,结果提示为TSC2基因c.4013-4014 delCA(p.Ser1338Cysfs)杂合突变。现精神运动发育尚可,学习成绩中上水平,身高体重均处于同龄人水平。先证者母亲,自幼面部可见对称性针尖样血管纤维瘤,右侧腰部见钙化星形细胞错构瘤(图 2B),前胸、双上肢散在分布小片状色素脱失斑,共4处。经家系一代测序验证,先证者母亲亦存在TSC2基因c.4013-4014 delCA杂合突变。先证者父亲、外祖父母均无TSC相关症状。

|
图 2 家系2先证者影像学特征和先证者母亲皮肤改变 A:先证者头颅CT检查示右侧侧脑室旁两个点状钙化灶(箭头所示);B:先证者母亲右侧腰部钙化星形细胞错构瘤。 |
1.2 外周血基因组DNA提取
在知情同意的前提下,取家系1、2先证者、先证者父母和家系2先证者外祖父母外周血2~3 mL,EDTA-K2抗凝,采用GentraPuregene Blood Kit(Qiagen公司生产)试剂盒提取基因组DNA,操作步骤按照说明书。检测DNA纯度和浓度(Nanodrop2000蛋白核酸分析仪),要求DNA纯度在1.8~2.0之间,浓度在20~100 ng/μL之间。
1.3 TSC相关基因高通量测序分析委托深圳华大临床检验中心将先证者DNA打断并制备文库,通过定制芯片对TSC1和TSC2目标基因的编码区及邻近剪切区的DNA进行捕获和富集,最后使用高通量测序平台进行突变检测。检测基因目标区长度约26 579 bp,目标区覆盖度95%以上,目标区平均深度509.58×,目标区平均深度>30×位点所占比例为100%。
1.4 引物设计NCBI查询TSC2基因序列(NG_005895),根据高通量测序筛选出的突变位点,采用Primer Premier 6.0设计引物并由上海生工公司合成,引物序列见表 1。
| 表 1 TSC2基因相应突变位点扩增引物 |
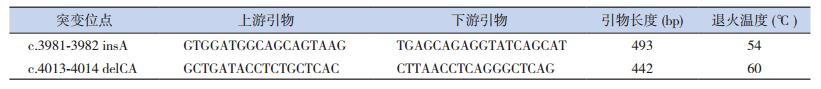
1.5 PCR扩增
应用立陶宛Thermo Scientific公司试剂,反应体系:ddH2O 15.5 μL,Buffer、Mg2+、dNTP各3 μL,正反引物各1 μL,Taq DNA酶1.5 μL,模板DNA 2 μL,总体系为30 μL。扩增条件:96℃ 3 min预变性;94℃ 1 min变性,退火1 min(相应退火温度见表 1),72℃延伸1 min,35个循环;72℃ 3 min,使PCR反应完全以提高扩增产量。取扩增产物5 μL,TaKaRa公司生产的6×Loading Buffer 1 μL,混匀,2%琼脂糖(Biowest Agarose)凝胶电泳以验证产物特异性。
1.6 PCR产物序列测定取PCR扩增产物25 μL,用表 1中PCR扩增引物作测序引物,测序委托上海生工公司完成。具体步骤如下:PCR产物纯化(生工公司纯化试剂);2%琼脂糖凝胶电泳确认其纯度和浓度(Biowest Agarose琼脂糖粉,生工公司的marker);根据PCR产物长度确定DNA模板浓度(长度为200~500 bp的模板用量为2~10 ng),DNA测序反应(ABI PCR仪,美国);产物上机前用酒精纯化;上测序仪(ABI 3730XL);采用Chromas软件分析数据。
2 结果家系1先证者检出TSC2基因c.3981-3982 insA(p.Asp1327AspfsX87)杂合突变(图 3),先证者父母该位点均未见异常。该框移突变导致氨基酸序列在第1413位提前终止编码,其致病性尚未见文献报道。家系2先证者、先证者母亲均检出c.4013-4014 delCA(p.Ser1338Cysfs)杂合突变(图 4),为母源性遗传,先证者父亲、先证者外祖父母该位点均未见异常。该框移突变导致氨基酸序列在第1412位提前终止编码,其致病性亦未见文献报道。

|
图 3 TSC2基因c.3981-3982 insA杂合突变测序结果 上图为正常对照,下图为先证者c.3981-3982 insA杂合突变,箭头所示为突变位点。 |

|
图 4 TSC2基因c.4013-4014 delCA杂合突变测序结果 上图为正常对照,下图为先证者c.4013-4014 delCA杂合突变,箭头所示为突变位点。 |
两突变在HGMD数据库(www.hgmd.cf.ac.uk/ ac/index.php)里未查出;在千人基因组计划数据库中检出频率为0;在深圳华大基因收集的大于200例本地正常人测序样本中的发生频率为A(发生频率0~0.01为A;0.01~0.05为B;0.05~1为C);用Mutationtaster软件(www.mutationtaster.org/)对其进行突变功能预测,预测分数均为1(预测结果分数范围为0~1,分数越高提示致病可能性越大)。
3 讨论TSC1编码的错构瘤蛋白和TSC2编码的马铃薯蛋白二者在细胞质内形成复合物,间接调控细胞增殖、细胞分裂和细胞黏附等,当任一基因发生突变时,胞内复合物形成受阻,对细胞增殖的抑制作用减弱或消失,造成细胞增殖过快而出现错构瘤损害[5]。TSC1-TSC2复合物在细胞中发挥作用是通过TSC2的GAP功能域(GTPase激活蛋白)间接激活哺乳动物雷帕霉素靶蛋白途径(mTOR),进而通过核糖体蛋白S6激酶(S6K)去磷酸化以达到抑制蛋白质合成、细胞生长、增殖和自我吞噬的作用[6]。因此,TSC1-TSC2复合物形成异常、TSC2基因突变造成GAP域异常和mTOR磷酸化水平异常,均可出现TSC相关临床表现。TSC2基因主要有两个功能域,即Hamartin结合域和GAP域[7],经Swiss-prot软件(www.uniprot.org/)在线查询可知GAP功能域位于第1531~1758位氨基酸,本研究讨论的TSC2基因c.3981-3982 insA(p.Asp1327AspfsX87)突变导致氨基酸序列在第1413位提前终止编码,c.4013-4014 delCA(p.Ser1338Cysfs)突变导致氨基酸序列在第1412位提前终止编码,二者均终止于GAP功能域前,导致GAP域全部缺失,为两家系的致病原因。
有不少国内外文献已报道错义突变、无义突变和框移突变导致GAP功能域异常,从而引发疾病。Mayer等[8]曾报道1例临床症状较轻的TSC,发现TSC2基因c.4684G>A(G1556S),此错义突变位于GAP域内,导致非极性的甘氨酸变成极性的丝氨酸,抑制S6K磷酸化的能力减弱,引起信号通路异常而致病。Chen等[9]在产前诊断中发现1例TSC2基因无义突变,即c.4830G>A(W1610X),此突变造成氨基酸编码发生提前终止,GAP功能域部分缺失,引起心脏横纹肌瘤和脑内异常等表现。Yu等[10]报道1例致病性的缺失突变TSC2基因c.2690delT(p.Phe897SerfsX947),此框移突变导致GAP功能域完全缺失,引起一系列临床症状。
本研究讨论的TSC2基因c.3981-3982 insA、c.4013-4014 delCA在HGMD数据库(www.hgmd.cf.ac.uk/ac/index.php)里未查出;在千人基因组计划数据库中检出频率为0;在深圳华大基因收集的大于200例本地正常人测序样本中的发生频率为A(发生频率0~0.01为A);用Mutationtaster软件(www.mutationtaster.org/)对其进行突变功能预测,预测分数均为1(预测结果分数范围为0~1,分数越高提示致病可能性越大)。经过文献复习、氨基酸结构分析,结合先证者临床表现等,推测这两个框移突变为新发的致病性突变,但是突变蛋白功能仍需要细胞模型和动物模型的进一步验证,突变体结构需要X衍射等技术的检测。
TSC2基因大,致病突变数量多,嵌合型比例高,基因检测非常复杂。明确基因突变有助于TSC家系遗传咨询和利用分子遗传学方法进行早期产前诊断。Curatolo等[11]在研究TSC基因型与表型的关系中阐明,TSC2突变引起的临床症状较TSC1更严重,如更易发生智力低下、癫癎、面部血管纤维瘤等。本研究中TSC2基因两突变先证者均有癫癎和面部血管纤维瘤表现,其中c.3981-3982 insA突变患儿临床表型严重,发病年龄较小,而c.4013-4014 delCA突变患儿表型较轻,智力发育正常。由于c.3981-3982 insA突变导致腺苷酸活化蛋白激酶(AMPK)[12]介导的磷酸化功能缺失,而c.4013-4014 delCA突变后此功能尚存在,因此推测这是TSC2基因c.3981-3982 insA突变较c.4013-4014 delCA表型严重的原因。
综上,本研究检出的TSC2基因c.3981-3982 insA、c.4013-4014 delCA突变为新发致病性框移突变,这两个新发突变的发现不仅丰富了基因突变谱,也为患病家系产前基因诊断提供了依据。
| [1] | Huang CH, Peng SS, Weng WC, et al. The relationship of neuroimaging findings and neuropsychiatric comorbidities in children with tuberous sclerosis complex[J]. J Formos Med Assoc, 2015, 114 (9): 849–854. DOI:10.1016/j.jfma.2014.02.008 |
| [2] | Ismail NF, Nik Abdul Malik NM, Mohseni J, et al. Two novel gross deletions of TSC2 in Malaysian patients with tuberous sclerosis complex and TSC2/PKD1 contiguous deletion syndrome[J]. Jpn J Clin Oncol, 2014, 44 (5): 506–511. DOI:10.1093/jjco/hyu024 |
| [3] | Kingswood JC, de Vries PJ. Tuberous sclerosis complex[J]. Paediatr Child Health, 2015, 25 (10): 467–473. DOI:10.1016/j.paed.2015.06.008 |
| [4] | Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations[J]. Hum Genet, 2007, 121 (3-4): 389–400. DOI:10.1007/s00439-006-0308-9 |
| [5] | De Waele L, Lagae L, Mekahli D. Tuberous sclerosis complex:the past and the future[J]. Pediatr Nephrol, 2015, 30 (10): 1771–1780. DOI:10.1007/s00467-014-3027-9 |
| [6] | Mazhab-Jafari MT, Marshall CB, Ho J, et al. Structure-guided mutation of the conserved G3-box glycine in Rheb generates a constitutively activated regulator of mammalian target of rapamycin (mTOR)[J]. J Biol Chem, 2014, 289 (18): 12195–12201. DOI:10.1074/jbc.C113.543736 |
| [7] | Astrinidis A, Henske EP. Tuberous sclerosis complex:linking growth and energy signaling pathways with human disease[J]. Oncogene, 2005, 24 (50): 7475–7481. DOI:10.1038/sj.onc.1209090 |
| [8] | Mayer K. Characterisation of a novel TSC2 missense mutation in the GAP related domain associated with minimal clinical manifestations of tuberous sclerosis[J]. J Med Genet, 2004, 41 (5): e64. DOI:10.1136/jmg.2003.010835 |
| [9] | Chen CP, Su YN, Hung CC, et al. Novel mutation in the TSC2 gene associated with prenatally diagnosed cardiac rhabdomyomas and cerebral tuberous sclerosis[J]. J Formos Med Assoc, 2006, 105 (7): 599–603. DOI:10.1016/S0929-6646(09)60157-1 |
| [10] | Yu Z, Zhang X, Guo H, et al. A novel TSC2 mutation in a Chinese family with tuberous sclerosis complex[J]. J Genet, 2014, 93 (1): 169–172. DOI:10.1007/s12041-014-0320-0 |
| [11] | Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex[J]. Semin Pediatr Neurol, 2015, 22 (4): 259–273. DOI:10.1016/j.spen.2015.10.002 |
| [12] | Liu L, Pan Y, Song Y, et al. Activation of AMPK α2 inhibits airway smooth muscle cells proliferation[J]. Eur J Pharmacol, 2016, 791 : 235–243. DOI:10.1016/j.ejphar.2016.09.003 |