 2017, Vol. 19
2017, Vol. 19



2. 河北医科大学公共卫生学院卫生检验教研室, 河北 石家庄 050017;
3. 四川大学华西第二医院产科, 四川 成都 610041;
4. 四川大学华西公共卫生学院卫生检验与检疫系, 四川 成都 610041
目前研究表明,肠道菌群参与人体的诸多生理功能,其结构与宿主的健康息息相关。生命早期建立正常的肠道菌群是今后健康生活的重要一步[1]。从婴儿出生开始,细菌逐渐在肠道内定植,1岁时基本形成类似于成人的肠道菌相,并在随后的1~2年内逐步完善[2]。最早定植的菌群对肠道生态系形成、长期的菌群构成以及肠道活动十分重要[3]。因此婴儿期是肠道菌群构建的重要时期。
肠道菌群数量庞大,约为1014个,超过1 000多个种类,其中60%~80%无法单独在体外培养[4]。早期研究依赖于培养方法认识和了解肠道菌群具有偏倚和不完整性。近年来,针对细菌16S rRNA核酸序列的现代分子生物学方法弥补了培养依赖方法的缺陷,为肠道菌群的研究提供了新的方法和思路[5]。变性梯度凝胶电泳(denatured gradient gel electrophoresis, DGGE)是分析环境系统中微生物群落的有效指纹图谱技术,近年已被用于肠道菌群的结构、多样性、动态变化的研究中[6]。
本研究通过PCR-DGGE分析婴儿期优势菌种构成,了解婴儿期肠道菌群菌种随时间变化的特点,探索婴儿期肠道菌群、特别是早期的定植规律。
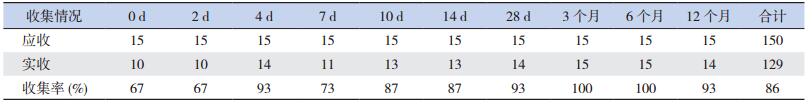
1 资料与方法 1.1 研究对象及粪便样品采集以2013年3~5月在四川大学华西妇产儿童医院出生的正常足月新生儿为研究对象。纳入标准:家庭居住在成都市主城区;胎龄满37周及以上,无任何畸形和疾病的活产新生儿;新生儿家长配合,愿意参加实验研究,并签署知情同意书。排除标准:新生儿因病理性黄疸、新生儿肺炎等疾病服用过抗生素或微生态制剂。本次共纳入31例新生儿,分别收集新生儿出生后第0、2、4、7、10、14、28天,以及婴儿期3、6、12月时的新鲜粪便样品,2 h内用冰盒转移至-80℃超低温冰箱冻存。随访期间有16例在婴儿期因感染性疾病使用微生态制剂或抗生素,予以排除。剩余15例婴儿,平均胎龄为39.0±1.0周,平均出生体重为3 218±221 g。其中男6例,女9例;顺产3例,剖腹产12例;母乳喂养9例,混合喂养(母乳为主)6例;辅食添加开始时间集中在婴儿出生后4~7个月,其中第4个月开始2例,第5个月开始5例,第6个月开始7例,第7个月开始1例;9例婴儿在随访期内断奶,6例婴儿在第12个月仍然有母乳喂养。粪便样品收集情况见表 1。
| 表 1 粪便样品收集情况 |
1.2 主要实验仪器及试剂
PCR仪、通用突变检测系统、凝胶成像系统、电泳仪均购自美国BIO-RAD公司;微量离心机(美国Thermo Fisher Scientific公司)。TIANamp stool DNA Kit(北京天根生化科技有限公司);2×Taq MasterMix(北京康为世纪科技有限公司);40%丙烯酰胺/甲叉双丙烯酰胺(37.5:1)、尿素、去离子甲酰胺(APS)、过硫酸铵、TEMED、SYBR Green Ⅰ、50×TAE均购自上海生工生物工程股份有限公司。
1.3 粪便基因组DNA提取及混合模板制备采用TIANamp stool DNA Kit提取每一个粪便样品的DNA,将各样品粪便基因组DNA按相同时间点进行等体积混合,获得婴儿出生后第0、2、4、7、10、14、28天,以及3、6、12月,10个时间点的混合基因组DNA。
1.4 粪便混合基因组16S rRNA PCR扩增选择16S rRNA V6-V8区通用引物(U968-GC-f/L1401-r)对粪便混合基因组DNA进行PCR扩增,扩增长度约为489 bp;U968-GC-f:5' -CG-CCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACG-GGGGGAACGCGAAGAACCTTAC-3' ;L1401-r:5' -CGGTGTGTACAAGACCC-3' ,由上海生工生物工程股份有限公司合成。反应体系:2×Taq Mastermix 25 μL,上、下游引物(10 μM)各2 μL,混合基因组DNA模板4 μL,补充ddH2O至50 μL。PCR反应条件:94℃预变性5 min;10个梯度退火温度循环(每个循环包括94℃变性45 s,退火温度从60℃降到54℃,每个循环降低0.6℃,时间1 min,72℃延伸1 min);25个恒定退火温度循环(每个循环包括94℃变性45 s,56℃退火45 s,72℃延伸1 min);72℃终延伸2 min。PCR产物经2.0%琼脂糖凝胶电泳验证后,凝胶成像系统拍照记录结果。
1.5 PCR产物的DGGE检测用DGGE对上述所得PCR扩增产物进行菌群结构的检测。DGGE反应条件:变性梯度凝胶8%,变性梯度40%~60%。电泳条件:1×TAE电泳缓冲液、60℃、200 V,电泳5 min后,70 V电泳16 h。电泳完毕后,用10 mL SYBR Green Ⅰ核酸染液避光染色15 min,染色3次后,去离子水洗去表面染液。凝胶成像系统拍照,导出TIFF格式图像待处理。
1.6 DGGE目的条带测序鉴定使用无菌手术刀切割DGGE图谱上亮度较高、代表优势菌的条带,将胶条用无菌去离子水漂洗2次后,10 μL去离子水4℃浸泡过夜。以浸出液作为PCR模板,进行16S rRNA V6-V8区PCR扩增。PCR产物用2%琼脂糖凝胶电泳验证后,送往上海生工生物工程股份有限公司行克隆测序,进行菌种水平鉴定。测序结果通过GenBank进行序列的同源性比对,选择Total Score最高且Ident指数大于97%者作为结果。
2 结果 2.1 婴儿期肠道菌群结构变迁分别将婴儿出生后10个时间点的混合粪便样本16S rRNA V6-V8区的PCR产物进行DGGE,获得DGGE指纹图谱(图 1)。由图中可以看出,各泳道间条带数量、位置及亮度上均存在不同程度的差异。婴儿期的肠道菌群结构处于不稳定状态,不同时间点婴儿的肠道菌群在其菌种组成上存在显著的差异。

|
图 1 婴儿期肠道菌群DGGE指纹图谱 图中1~24条带较亮,代表优势菌种,作为目的条带切割下来,进行克隆测序。 |
2.2 婴儿期肠道菌群菌种构成
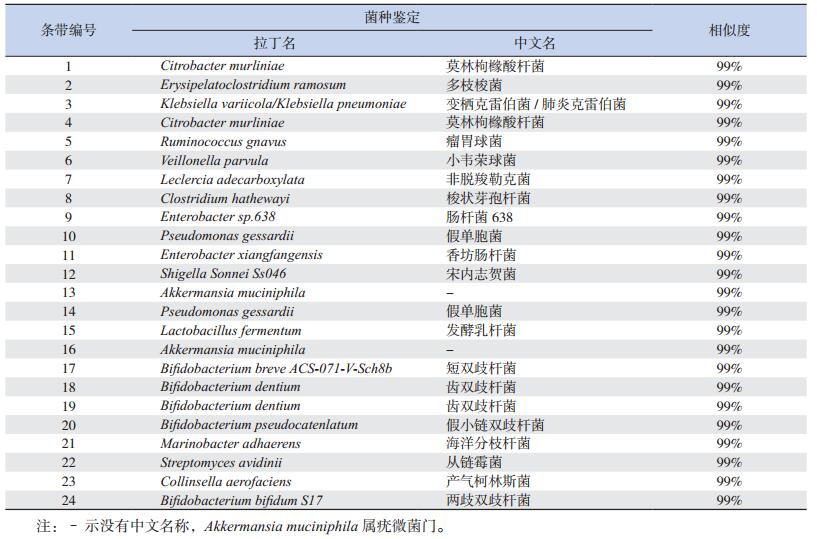
DGGE指纹图谱中目的条带菌种测序鉴定结果见表 2。从细菌对氧气的需求进行细菌分类,莫林枸橼酸杆菌、非脱羧勒克菌、肠杆菌、假单胞菌、宋内志贺菌、海洋分枝杆菌、从链霉菌属于专性需氧菌,发酵乳杆菌和多枝梭菌属于兼性厌氧菌,而瘤胃球菌、小韦荣球菌、梭状芽孢杆菌、Akkermansia muciniphila、双歧杆菌属于专性厌氧菌。从生物分类学角度看,莫林枸橼酸杆菌、克雷伯菌、非脱羧勒克菌、肠杆菌、假单胞菌、宋内志贺菌、海洋分枝杆菌属于变形菌门,瘤胃球菌、多枝梭菌、小韦荣球菌、梭状芽孢杆菌、发酵乳杆菌属于厚壁菌门,Akkermansia muciniphila属于疣微菌门,双歧杆菌、产气柯林斯菌、从链霉菌属于放线菌门。
| 表 2 目的条带的菌种鉴定 |
2.3 婴儿期肠道菌群菌种演替
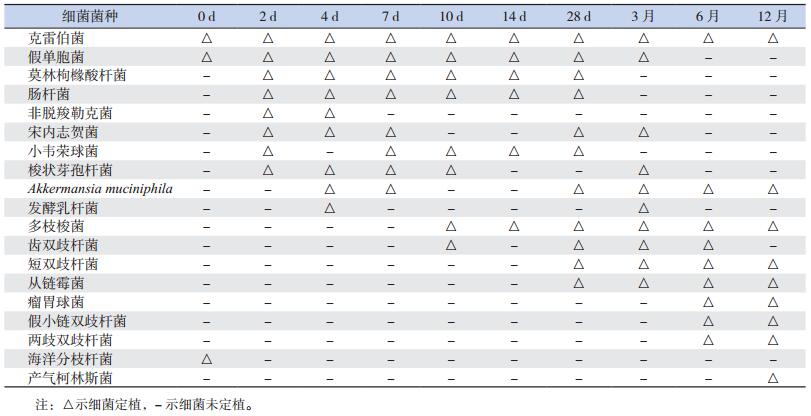
结合图 1及表 2,分析婴儿期肠道菌群定植演替过程(表 3)。婴儿肠道内变形菌门的细菌定植最早,以专性需氧菌肠杆菌、假单胞菌为主,紧接着厚壁菌门专性厌氧菌梭状芽孢杆菌、小韦荣球菌出现定植,随后专性厌氧的疣微菌门Akkermansia muciniphila和厚壁菌门的兼性厌氧菌发酵乳杆菌、多枝梭菌开始定植,放线菌门细菌定植最晚,以双歧杆菌为主,并逐渐成为婴儿肠道内的优势菌群。肠道内细菌定植演替规律为:从细菌对氧气的需求来看,婴儿肠道内最早定植的是专性需氧菌,然后是兼性厌氧菌和专性厌氧菌。从生物分类学的角度来看,婴儿肠道内变形菌门细菌定植最早,厚壁菌门细菌次之,随后是疣微菌门细菌,放线菌门细菌定植最晚。婴儿出生第28天后,变形菌门细菌数量开始减少并逐渐消失,而以双歧杆菌为主的放线菌门细菌大量增殖,成为婴儿肠道内的优势菌群。
| 表 3 婴儿期肠道菌群定植演替 |
3 讨论
早期的肠道菌群研究依赖于培养的方法。研究结果表明,婴儿肠道内最初定植的细菌主要为葡萄球菌、链球菌、肠球菌、肠杆菌等厌氧菌或兼性厌氧菌,随着厌氧菌或兼性厌氧菌消耗氧气形成厌氧环境,双歧杆菌、拟杆菌和梭菌等专性厌氧菌的数量增加[7]。尤其是放线菌中双歧杆菌大量定植,成为婴儿肠道内的优势菌群,这与本研究中结果基本一致,以细菌16S rRNA核酸序列的现代分子生物学方法亦证实了这一点[8]。培养依赖方法耗时耗力、烦杂、再现性差,以及选择培养基本身存在的缺陷,造成检出微生物种群偏倚和检出菌株数据偏低等,因此迄今为止依赖于培养方法的婴儿肠道菌群研究还未能全面、客观地反映出婴儿期肠道菌群菌种演替规律,现代分子生物学方法逐渐成为研究的主流方式[6]。
现代分子生物学技术可同时检测放线菌门、厚壁菌门、拟杆菌门、变形菌门、疣微菌门下的多种细菌,揭示了婴儿期大约30%培养依赖方法所未能鉴定的菌种,如梭状芽胞杆菌和瘤胃球菌属等[3, 8]。Bäckhed等[9]用宏基因组鸟枪法测序对98名婴儿及其母亲肠道菌群进行了更为全面的分析,发现厚壁菌门和拟杆菌门是最常见的菌门,其次是放线菌门和变形菌门。具体来看,新生儿的标志菌群是大肠埃希菌/志贺菌属、链球菌属、罗氏菌属,4月龄婴儿标志菌群为双歧杆菌属、乳酸杆菌属、颗粒链菌属、柯林斯菌属、韦荣球菌属,12月龄婴儿标志菌群包括新生儿出现的拟杆菌,4月龄时出现的放线菌、厌氧球菌和梭菌,以及刚出现的埃肯菌属等。Lee等[10]用测序方法对20名韩国4周龄婴儿的肠道菌群进行分析,放线菌门、厚壁菌门和变形菌门是主要菌群,其中长双歧杆菌、唾液链球菌、乳链球菌、假肺炎链球菌、格氏乳酸杆菌是常见菌种。本研究婴儿期主要菌门是变形菌门、厚壁菌门、放线菌门和疣微菌门,未检出拟杆菌门细菌,优势菌种与前二者相比有相同之处,但总体来说差异比较大。
不同研究结果之间的差异较大,可能与研究人群、研究方法有关。Fallani等[11]对欧洲6周龄的婴儿用荧光原位杂交(FISH)分析,结果表明双歧杆菌、拟杆菌和肠球菌是其主要的优势菌种,但北欧国家婴儿粪便中的双歧杆菌比例更高,南欧国家拟杆菌的多样性较高。可见不同地域婴儿的肠道菌群具有其各自的特点,因此对我国婴儿肠道菌群研究是必要的。现代分子生物学技术,如测序、荧光原位杂交、实时荧光定量PCR等,虽然独立、快速、高效,但也受到了16S rRNA基因序列的限制,存在分辨率较低的问题[12]。探针或所测定16S rRNA序列的长短,一定程度影响检出细菌的多少,而不同研究方法在这一点上往往是有差异的。宏基因组方法分辨率相对较高,具有高通量、信息全面、系统识别等特点,成为当前肠道菌群的重要分析方式,但价格也较为昂贵、难进行多次重复。同时宏基因组方法在通常使用上主要集中在微生物分类学菌属水平以上肠内细菌的测定,在菌种水平的测定上存在着信赖度较差的问题,特别是在婴儿早期的肠道菌群构成简单、优势菌群种类及数量均少于成人的情况下,作为初探宏基因组方法未必是最合适的方法。相反,本研究采用的PCR-DGGE方法相对而言操作更加简单、快速、经济,因此选用PCR-DGGE方法初步分析婴儿期肠道菌群菌种演替规律。另外,婴儿肠道菌群定植过程受到诸多因素影响,比如生产方式、喂养方式、抗生素的使用、家庭构成和家庭环境等[8]。本研究虽然对婴儿生产方式、喂养方式、辅食添加等追踪调查,但同样受到小样本量的限制,难以分组控制上述因素对婴儿肠道菌群的可能影响,而将15名婴儿粪便样品按等时间等体积混合作为一个整体分析,初步探索健康婴儿在婴儿期的菌种演替规律,为后续的研究提供基础数据。
目前大多数研究认为,胎儿在母体时肠道内呈无菌状态,出生后暴露于细菌环境中开始定植过程,最早始于婴儿的分娩,而目前有少数研究采用分子生物学方法在羊水、子宫、胎盘内检测到细菌的存在[13-14],本研究中生后胎便里观察到有细菌存在,提示肠道菌群定植的始点可能更早。相关研究表明,厚壁菌门中的瘤胃球菌属在婴儿肠道早期的出现频率很高[15],且相比牛乳喂养婴儿,母乳喂养和山羊奶喂养婴儿肠道中的厚壁菌门几乎是单一瘤胃球菌属[16]。本研究中瘤胃球菌出现时间相对较晚,但从指纹图谱可以看出它在厚壁菌门中处于优势状态,其在婴儿肠道中扮演的角色有待进一步研究。疣微菌门中的Akkermansia muciniphila是新发现能特异地生长在人肠道黏膜层内,对人体健康有益的肠道细菌之一[17]。近年的研究表明,Akkermansia muciniphila能影响人体物质和能量代谢,动物实验发现它具有抑制肥胖、抗炎症和改善糖尿病等代谢性疾病的可能性[18]。在本研究中,Akkermansia muciniphila作为婴儿期优势菌种比双歧杆菌更早地被发现在受试婴儿肠道内,其对婴儿健康的影响值得今后更深入的研究。
已有研究结果表明,不论是培养依赖方法,还是分子生物学方法,都说明了婴儿早期肠道菌群的定植是一个“混乱”的过程,呈现出一个高动态、不稳定和低多样性的特点[12]。尽管如此,婴儿期肠道菌群的定植和发展所知相对较少[9]。本研究设计了前密后疏的检测时间点,真实有效地反映了婴儿期肠道菌群定植演替过程,加深了对婴儿期肠道菌群定植的认识,为继续深入研究婴儿期肠道菌群定植规律提供了宝贵的基础数据。
| [1] | Echarri PP, Graciá CM, Berruezo GR, et al. Assessment of intestinal microbiota of full-term breast-fed infants from two different geographical locations[J]. Early Hum Dev, 2011, 87 (7): 511–513. DOI:10.1016/j.earlhumdev.2011.03.013 |
| [2] | Matamoros S, Gras-Leguen C, Le Vacon F, et al. Development of intestinal microbiota in infants and its impact on health[J]. Trends Microbiol, 2013, 21 (4): 167–173. DOI:10.1016/j.tim.2012.12.001 |
| [3] | Scholtens PA, Oozeer R, Martin R, et al. The early settlers:intestinal microbiology in early life[J]. Annu Rev Food Sci Technol, 2012, 3 : 425–447. DOI:10.1146/annurev-food-022811-101120 |
| [4] | Dave M, Higgins PD, Middha S, et al. The human gut microbiome:current knowledge, challenges, and future directions[J]. Transl Res, 2012, 160 (4): 246–257. DOI:10.1016/j.trsl.2012.05.003 |
| [5] | Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease[J]. Nature, 2007, 449 (7164): 811–818. DOI:10.1038/nature06245 |
| [6] | Ruengsomwong S, Korenori Y, Sakamoto N, et al. Senior Thai fecal microbiota comparison between vegetarians and non-vegetarians using PCR-DGGE and real-time PCR[J]. J Microbiol Biotechnol, 2014, 24 (8): 1026–1033. DOI:10.4014/jmb.1310.10043 |
| [7] | Inoue Y, Shimojo N. Microbiome/microbiota and allergies[J]. Semin Immunopathol, 2015, 37 (1): 57–64. DOI:10.1007/s00281-014-0453-5 |
| [8] | Adlerberth I, Wold AE. Establishment of the gut microbiota in Western infants[J]. Acta Paediatr, 2009, 98 (2): 229–238. DOI:10.1111/j.1651-2227.2008.01060.x |
| [9] | Bäckhed F, Roswall J, Peng Y, et al. Dynamics and stabilization of the human gut microbiome during the first year of life[J]. Cell Host Microbe, 2015, 17 (5): 690–703. DOI:10.1016/j.chom.2015.04.004 |
| [10] | Lee SA, Lim JY, Kim BS, et al. Comparison of the gut microbiota profile in breast-fed and formula-fed Korean infants using pyrosequencing[J]. Nutr Res Pract, 2015, 9 (3): 242–248. DOI:10.4162/nrp.2015.9.3.242 |
| [11] | Fallani M, Young D, Scott J, et al. Intestinal microbiota of 6-week-old infants across Europe:geographic influence beyond delivery mode, breast-feeding, and antibiotics[J]. J Pediatr Gastroenterol Nutr, 2010, 51 (1): 77–84. DOI:10.1097/MPG.0b013e3181d1b11e |
| [12] | O'Toole PW, Claesson MJ. Gut microbiota:changes throughout the lifespan from infancy to elderly[J]. Int Dairy J, 2010, 20 (4): 281–291. DOI:10.1016/j.idairyj.2009.11.010 |
| [13] | Azad MB, Konya T, Maughan H, et al. Gut microbiota of healthy Canadian infants:profiles by mode of delivery and infant diet at 4 months[J]. CMAJ, 2013, 185 (5): 385–394. DOI:10.1503/cmaj.121189 |
| [14] | Ardissone AN, de la Cruz DM, Davis-Richardson AG, et al. Meconium microbiome analysis identifies bacteria correlated with premature birth[J]. PLoS One, 2014, 9 (3): e90784. DOI:10.1371/journal.pone.0090784 |
| [15] | Sagheddu V, Patrone V, Miragoli F, et al. Infant early gut colonization by lachnospiraceae:high frequency of ruminococcus gnavus[J]. Front Pediatr, 2016, 4 : 57. |
| [16] | Tannock GW, Lawley B, Munro K, et al. Comparison of the compositions of the stool microbiotas of infants fed goat milk formula, cow milk-based formula, or breast milk[J]. Appl Environ Microbiol, 2013, 79 (9): 3040–3048. DOI:10.1128/AEM.03910-12 |
| [17] | Everard A, Belzer C, Geurts L, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity[J]. Proc Natl Acad Sci U S A, 2013, 110 (22): 9066–9071. DOI:10.1073/pnas.1219451110 |
| [18] | Caesar R, Tremaroli V, Kovatcheva-Datchary P, et al. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling[J]. Cell Metab, 2015, 22 (4): 658–668. DOI:10.1016/j.cmet.2015.07.026 |