 2017, Vol. 19
2017, Vol. 19

2. 中山大学孙逸仙纪念医院儿科, 广东 广州 510120

 , WANG Hai-Yan, JIANG Xiao-Yun, MO Ying, YUE Zhi-Hui, CHEN Hua-Mu, LIU Ting, LIN Hong-Rong
, WANG Hai-Yan, JIANG Xiao-Yun, MO Ying, YUE Zhi-Hui, CHEN Hua-Mu, LIU Ting, LIN Hong-Rong
Alport综合征(Alport syndrome, AS)是儿童最常见遗传性进行性肾炎,是由于肾小球基底膜(glomerular basement membrane, GBM)Ⅳ的胶原α3、α4或α5链(COL4A3、COL4A4或COL4A5)遗传缺陷所致[1]。包括3种遗传类型:X-连锁显性遗传AS(XL-AS)为COL4A5基因缺陷,临床最常见,约占所有AS患者的85%;常染色体隐性遗传AS(AR-AS)为COL4A3或COL4A4基因缺陷,约占AS患者的15%;常染色体显性遗传AS,临床罕见[2-4]。AS主要表现为血尿,感音神经性耳聋,眼部异常和进行性肾功能减退。早期表现轻微,仅为镜下血尿,伴或不伴蛋白尿,肾功能不全多在青春期后出现,听力和视力损害也出现较晚。由于AS儿童时期表现多不典型,缺乏特异性,临床易于误诊。本研究总结确诊的91例AS患儿的临床病理特征和诊治情况,以提高对AS的认识,减少诊治延误。
1 资料与方法 1.1 研究对象收集中山大学附属第一医院儿科2000年6月至2016年5月确诊的91例AS患儿的临床资料,并进行回顾性分析。其中男71例、女20例,中位数发病年龄为48(3~153)个月。
AS诊断参照美国学者提出的诊断标准[5]:(1)血尿家族史,家族中有男性患者进展至终末期肾病;(2)电镜下特征性的GBM增厚和分层;(3)进行性、高频区、感音神经性耳聋;(4)前锥形晶状体和黄斑周围斑点;(5)COL4A3、COL4A4或COL4A5在基底膜异常表达;(6)编码COL4A3、COL4A4或COL4A5的基因突变。符合以上6条标准中的2条即可诊断为AS。本研究以电镜下特征性的GBM改变,皮肤基底膜COL4A5或肾小球基底膜COL4A3、COL4A4或COL4A5表达缺陷,以及COL4A3、COL4A4或COL4A5基因缺陷为AS确诊依据。
1.2 临床资料收集收集AS患儿临床资料,包括起病年龄、发病至确诊的时间、血尿、蛋白尿、肾功能、肾外症状、家族史、有无接受激素和免疫抑制剂治疗等。收集肾脏病理资料,包括光镜、免疫荧光(IgM、IgG、IgA、Fg、C3、C1q的检测情况)、Ⅳ型胶原检测、电镜检查结果。部分患者留取外周血进行基因检测,应用靶基因芯片捕获高通量测序进行检测,阳性结果再用Sanger测序或实时定量PCR验证,由北京迈基诺基因科技有限公司、深圳华大临床检验中心完成。
1.3 统计学分析采用SPSS 20.0统计学软件进行数据处理,非正态分布的计量资料采用采用中位数(范围)表示,计数资料用百分率(%)表示。
2 结果 2.1 一般资料确诊的91例AS患儿中男71例、女20例。中位发病年龄为48(3~153)个月,0~3岁的55例(60%);4~6岁的20例(22%);7~13岁的16例(18%)。2000年6月至2008年5月诊断的19例AS患儿通过电镜确诊。2008年6月以后的63例进行了肾脏Ⅳ型胶原α3和α5链免疫荧光检测,58例确诊为AS(1例同时经基因确诊)、2例通过电镜确诊,3例通过基因确诊。4例外院肾活检未能确诊的患儿中1例通过皮肤基底膜Ⅳ胶原α5链免疫荧光、3例通过基因检测确诊。5例未行肾活检的患儿通过基因检测确诊。
2.2 临床特点91例患儿均有血尿(100%),其中孤立性血尿5例(5%);血尿伴间歇性蛋白尿32例(35%),血尿伴持续性蛋白尿33例(36%);肾病综合征(肾炎型)21例(23%)。9例(10%)患儿出现肾功能损害,肾功能损害出现年龄为132(96~156)个月,男7例、女2例,慢性肾脏病Ⅱ期4例、Ⅲ期3例、Ⅳ期1例、Ⅴ期1例,估算肾小球滤过率(eGFR)为50.4(4.9~79.9)mL/(min · 1.73m2)。
17例(19%)出现听力损害,均为轻-中度感音性神经性耳聋,男14例、女3例,中位年龄95(26~156)个月。其中14例无自觉症状,仅高频区轻中度的听力下降;3例有听力下降,均为男性,其中1例在12岁出现听力下降,2例在9岁出现。确诊眼部病变的19例(21%),男14例、女5例,中位年龄115(54~156)个月,8例为糖皮质激素相关性白内障、9例屈光不正、1例虹膜错构瘤、1例黄斑病变。
86例XL-AS患儿来自85个家系,其中61例(61/86,71%)XL-AS患儿有阳性家族史,父母一方及相关亲属表现为血尿,血尿伴蛋白尿,或慢性/隐匿性肾炎,肾功能不全,伴或不伴视力或听力异常。但这些患者在本中心就诊前,仅31例采集到家族史,6个家系确诊了AS。5例AR-AS患者中,4例有阳性家族史。
2.3 肾脏病理特点入院后82例患儿行肾活检。系膜无明显增生8例(10%),轻度增生68例(83%),轻-中度增生6例(7%);伴有球性硬化和/或节段性硬化25例(30%);节段球囊壁粘连4例(5%);内皮细胞增生11例(13%);小灶状间质损伤20例(32%)。免疫荧光全部阴性的34例(41%);48例可见少量免疫复合物系膜区沉积,其中IgM ±~++的30例(37%);Fg +的20例(24%);IgA± ~+ 7例(9%);IgG + 8例(10%);C3 + 9例(11%);C1q ±~++ 5例(6%)。电镜提示GBM厚薄不均、节段或广泛性撕裂、分层的53例(68%);GBM仅部分节段变薄或增厚21例(27%);GBM正常4例(5%);无肾小球4例。
63例进行了肾组织Ⅳ型胶原α3、α5链免疫荧光检测,53例α3和α5链在GBM、球囊壁和肾小管基底膜完全缺失或节段(弱)阳性,符合XL-AS;5例患儿α3链在GBM、包氏囊不表达,α5链在GBM不表达,在包氏囊壁或皮肤正常表达或稍减弱,符合AR-AS;1例男性患儿的α3、α5链表达不典型,表达于肾小球及肾小管基底膜、呈节段性减弱;4例α3、α5链表达正常。α3、α5链表达正常及不典型的5例患儿分别通过电镜(2例)或基因检测(3例)确诊。
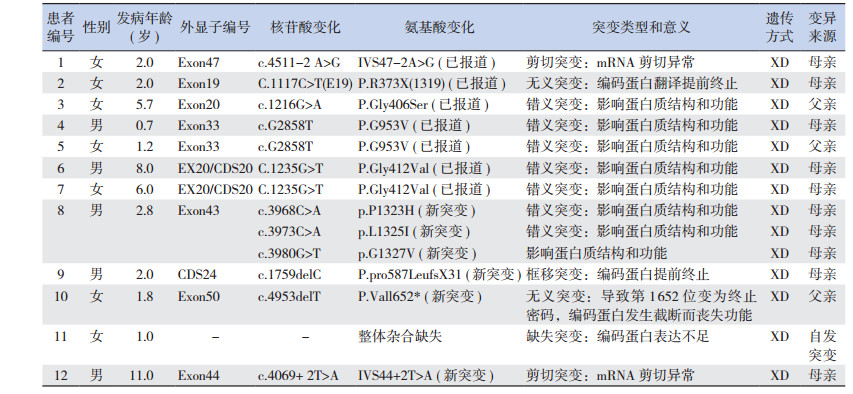
2.4 基因检测12例患儿经基因检测确诊,均为XL-AS,共发现6个COL4A5基因新突变位点:c.3968C > A,c.3973C > A,c.3980G > T,c.1759delC,c.4953delT,c.4069+ 2T > A,见表 1。其中1例患儿同时有3个COL4A5新突变位点,均来自母亲(图 1);1例患儿为COL4A5整体杂合缺失,为自发突变,父母均正常(图 2)。
| 表 1 12例基因诊断的AS患儿COL4A5基因突变情况 |

|
图 1 1例Alport综合征患者及其母亲的3个COL4A5基因突变 患儿检出c.3968C > A,c.3973C > A,c.3980G > T突变,患儿母亲为这些突变的杂合子。突变位点如箭头所示。 |

|
图 2 COL4A5基因整体杂合缺失患者的二代测序(A)和实时定量PCR验证(B) 红色椭圆形部分提示COL4A5基因整体杂合缺失。P1~7、FP1~7和MP1~7分别为患者或其父、母对应的COL4A5片段:X染色体107840607-107840798、107841932-107842100、107863489-107863656、107930712-107930924、107930712-107930924、107938497-107938669、107938497-107938669;C1、C2和C3为正常男性对照样本。P1~7提示患儿COL4A5链扩增整体杂合缺失;FP1~7提示患者父亲为半合子;MP1~7提示患儿母亲COL4A5双链正常扩增。 |
2.5 误诊情况
91例患儿,起病至确诊的中位时间为2年3个月(1周~12年),45例(45/91,49%)在确诊前误诊,误诊中位时间为2年5个月(1周~11年),其中男35例、女10例。确诊前诊断孤立性血尿,急性、迁延性、慢性肾小球肾炎,肾病综合征,IgA肾病或未确诊,但未使用激素或免疫抑制剂治疗者,本研究不纳入误诊。确诊前41例(41/45,91%)接受了糖皮质激素和/或免疫抑制剂等治疗。
误诊为IgA肾病的8例,确诊前均使用了糖皮质激素和/或免疫抑制剂;其中3例肾活检提示轻度系膜增生,2例伴局灶节段性肾小球硬化、IgA弥漫系膜区沉积。最终,2例通过电镜确诊、6例通过Ⅳ型胶原检测确诊。
误诊为急性、迁延性、慢性肾小球肾炎的9例,确诊前均接受了糖皮质激素和/或免疫抑制剂治疗,6例有血尿、肾炎或肾功能不全的家族史,其中1例外院行肾活检,未行Ⅳ型胶原检测,电镜提示薄基底膜病可能性大。最终,4例经电镜确诊,4例通过Ⅳ型胶原检测确诊,1例通过基因确诊。
误诊为特发性肾病综合征的18例,其中1例于确诊前2个月(9岁)自觉听力下降。2例患儿外院肾活检均未行Ⅳ型胶原检测,1例光镜示系膜增生性肾小球肾炎,另1例光镜示局灶性节段性肾小球硬化。所有患儿确诊前均接受了糖皮质激素治疗,17例激素耐药,1例治疗后蛋白尿缓解、感染后多次复发;11例患儿联用了免疫抑制剂治疗。最终,6例经电镜确诊,7例经肾组织Ⅳ型胶原检测确诊,5例通过基因检测确诊。
误诊为紫癜性肾炎1例,无家族史及眼、耳病变,糖皮质激素或免疫抑制剂治疗无效,最终通过电镜确诊。
2例男性患儿误诊为乙型病毒性肝炎相关性肾炎,1例于确诊前3年11个月出现听力下降(中重度感音神经性耳聋),母亲有血尿、蛋白尿,糖皮质激素或免疫抑制剂治疗无效;2例肾活检Ⅳ型胶原检测提示AR-AS和XL-AS,电镜特征性GBM改变,肾组织HBsAg、HBeAg、HBcAg均阴性。
误诊为系膜增殖性肾小球肾炎的3例均无眼、耳病变,糖皮质激素和/或免疫抑制剂治疗效果不佳,其中2例外院肾活检提示MsPGN、均无免疫球蛋白沉积。最终,2例通过肾组织Ⅳ型胶原检测及电镜确诊,1例经皮肤活检确诊。
3 讨论AS早期表现缺乏特异性,诊断较为困难。在开展皮肤基底膜COL4 α5链和肾组织基底膜COL4 α3/4/5链免疫检测以及COL4 A3/4/5基因检测之前,确诊只能依靠电镜下特征性GBM超微结构改变。本研究结果显示,本区域AS患儿的误诊率仍较高,达49%,误诊时间较长,误诊中位时间2年3个月(1周至11年)。此外,91%(41/45)的被误诊患儿接受了不恰当的激素和/或免疫抑制剂治疗。虽然他克莫司和环孢素A对AS蛋白尿有效,但没有确诊AS前使用仍视为不恰当。
AS早期表现主要是血尿,随疾病进展,逐渐出现蛋白尿。少数可表现为肾病综合征,最易于误诊。本研究21例AS表现为肾病综合征,被误诊为特发性肾病综合征的18例,乙肝相关性肾炎1例,紫癜性肾炎1例。
AS患者可伴有听力和视力损害,以XL-AS男性患者发生较高[6]。听力损害主要表现为高频音域感音神经性耳聋或听力下降。视力损害典型表现为前圆锥形晶状体和黄斑周围斑点状变性[4]。AS患者听力和视力损害出现较晚,听力损害多在10岁以后出现,视力损害多在20岁以后出现[7]。因此,儿童眼耳病变的检出率远低于成人[8]。本研究17例患儿发现听力损害,但仅3例有自觉症状。19例患儿检测到眼部病变,其中1例为黄斑病变,其它均为非AS综合征相关的眼部改变。
大多数XL-AS患者(约80%)有血尿或肾功能衰竭家族史[9]。本研究71%(61/86)的XL-AS患者有血尿或肾功能衰竭家族史。但这些患者在本中心就诊前,仅31例采集到家族史,其中6个家系确诊AS。提示很多医生对本病仍缺乏警惕和认识。
导致AS患者误诊的另一个重要原因是,AS患者的肾脏病理早期肾小球多无明显改变,部分在5岁以后肾小球可见局灶轻微系膜增生,内皮损伤等轻微病变,进而出现节段或球性硬化,间质纤维化等,但均无特异性。本研究绝大多数肾活检患儿(90%,74/82)肾小球有轻度或中度系膜增生,35%有球性或节段硬化,或节段球囊壁粘连,13%有内皮增生;这些改变导致患者被误诊为增生性或硬化性肾小球疾病。
AS患者肾小球免疫荧光检测多阴性或仅有少量非特异性免疫复合物沉积,如IgM或Fg等,少数患者可有少量IgA或IgG沉积,应注意与IgA肾病、感染后肾小球肾炎等鉴别。本组7例患者有少量IgA沉积,其中3例被误诊为IgA肾病。
在皮肤和肾组织Ⅳ型胶原免疫检测和基因检测应用于AS诊断之前,电镜是确诊AS的唯一手段,特征性改变是GBM厚薄不均、分层、撕裂[10],但需疾病进展到一定程度才出现,因此早期容易出现漏诊或误诊。本中心开展肾组织/皮肤Ⅳ型胶原α链检测8年期间共诊断63例AS,仅53例(68%)电镜呈现特征性GBM改变。随着检测技术的发展,近年来基因检测已越来越多的应用于AS诊断。本研究12例患儿通过基因检测确诊,肾组织COL4 α3/α5链正常表达的4例有3例通过基因检测明确了诊断,还发现了6个COL4A5基因新突变位点。
总之,AS早期表现缺乏特异性,对于临床表现为持续肾小球性血尿或伴蛋白尿的患者应积极询问家族史,必要时行皮肤或肾组织COL4 α3/4/5链检测,注意GBM电镜改变,或行基因检测,以减少诊治延误。
| [1] | Hudson BG, Tryggvason K, Sundaramoorthy M, et al. Alport's syndrome, Goodpasture's syndrome, and type Ⅳ collagen[J]. N Engl J Med, 2003, 348 (25): 2543–2556. DOI:10.1056/NEJMra022296 |
| [2] | 丁洁, 张琰琴. Alport综合征精准诊治进展[J]. 中华肾病研究电子杂志, 2016, 5 (2): 53–55. |
| [3] | Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy[J]. J Am Soc Nephrol, 2013, 24 (3): 364–375. DOI:10.1681/ASN.2012020148 |
| [4] | Savige J. Alport syndrome:its effects on the glomerular filtration barrier and implications for future treatment[J]. J Physiol, 2014, 592 (Pt 18): 4013–4023. |
| [5] | Hanson H, Storey H, Pagan J, et al. The value of clinical criteria in identifying patients with X-linked Alport syndrome[J]. Clin J Am Soc Nephrol, 2011, 6 (1): 198–203. DOI:10.2215/CJN.00200110 |
| [6] | Alves FR, de A Quintanilha Ribeiro F. Revision about hearing loss in the Alport's syndrome, analyzing the clinical, genetic and bio-molecular aspects[J]. Braz J Otorhinolaryngol, 2005, 71 (6): 813–819. DOI:10.1016/S1808-8694(15)31253-2 |
| [7] | 罗岩, 胡六梅, 李维业. Alport综合征临床表现[J]. 国际眼科杂志, 2008, 3 (8): 618–619. |
| [8] | 何旭, 刘光陵, 夏正坤, 等. 47例Alport综合征临床与病理分析[J]. 中华儿科杂志, 2008, 46 (12): 914–918. DOI:10.3321/j.issn:0578-1310.2008.12.008 |
| [9] | 丁洁, 王云峰. Alport综合征诊断中应注意的几个问题[J]. 中国医刊, 2005, 40 (3): 21–23. |
| [10] | Heidet L, Gubler MC. The renal lesions of Alport syndrome[J]. J Am Soc Nephrol, 2009, 20 (6): 1210–1215. DOI:10.1681/ASN.2008090984 |