 2017, Vol. 19
2017, Vol. 19



2. 南京医科大学第二附属医院儿科, 江苏 南京 210003;
3. 南京医科大学第一附属医院生殖遗传检测中心, 江苏 南京 210029;
4. 南京大学附属南京鼓楼医院病理科, 江苏 南京 210008
进行性肌营养不良(progressive muscular dystrophy, PMD)是一种遗传性骨骼肌进行性无力和萎缩,共分为6型,其中以杜氏肌营养不良(Duchenne muscular dystrophy, DMD)最多见。DMD亦称假性肥大型肌营养不良,是肌营养不良蛋白dystrophin基因突变所致的一组X连锁隐性遗传病,通常男性发病,5岁前隐匿起病,进行性加重,多于20岁前死于心肺功能衰竭,是一种最严重的、致死性神经肌肉疾病。国内外关于DMD的CK变化规律,以及脐血间充质干细胞移植治疗的报道较少,本文章收集了6例DMD患者的临床资料,分析总结DMD的一般临床特点,长期随访揭示CK等酶谱的变化规律,对于进行脐血间充质干细胞移植的病例进行分析总结,为DMD的早期诊断及有效治疗措施的探索提供理论基础。
1 资料与方法 1.1 研究对象收集2010年1月至2015年10月南京医科大学第一附属医院诊治的6例DMD患者临床资料,均通过外周血DMD基因检测确诊。对患儿的症状,以及性别、年龄、血清酶谱、DMD基因检测、肌电图、肌肉活检和家族史等临床资料进行归纳总结。
1.2 DMD基因检测取EDTA抗凝外周静脉血。2010~2013年期间的病例采用mPCR+聚丙烯酰胺凝胶电泳法,针对导致DMD发病最常见的18个外显子(Pm、3、4、6、8、12、13、17、19、43、44、45、47、48、50、51、52、60)缺失突变进行检测;2014~ 2015年期间的病例采用多重连接探针扩增法(MLPA),对DMD基因79个外显子缺失和重复突变进行检测。
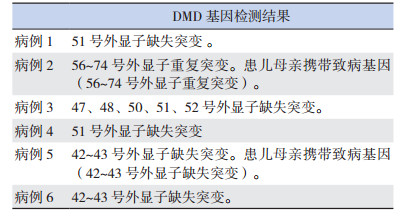
2 结果 2.1 一般资料6例DMD患儿均为男性,确诊年龄为1.2~11.5岁,平均4.3岁(4.3±3.6岁),均无家族史。患儿起病隐匿,均可独立行走,其中1例因肌酶异常就诊,2例因呼吸道感染就诊,3例因运动能力异常就诊。血清酶谱示谷丙转氨酶(ALT)、谷草转氨酶(AST)、乳酸脱氢酶(LDH)、α-羟丁酸脱氢酶(HBDH)、肌酸激酶(CK)、肌酸激酶同工酶(CKMB)均有同步性升高,以CK升高最显著、为正常的3.7~107.2倍(平均64±43倍)。病例3因呼吸道感染就诊时发现CK明显增高,于1.7岁行基因检查确诊,自确诊后至6.2岁运动功能一直正常,期间10次血生化检查提示血清酶谱呈波浪式进展,以CK的改变最显著(见图 1)。6例患儿于我院行DMD基因检测:1例为重复突变,5例为缺失突变,见表 1。病例2、病例5的母亲进行了外周血DMD基因检测,基因突变类型与患者一致(见图 2)。

|
图 1 病例3血清酶学的动态变化 |
| 表 1 6例DMD患者的基因检测结果 |

|
图 2 病例2及其母亲DMD基因的MLPA检测结果 患儿P034探针(A)检出外显子61-70重复突变,P035探针(B)检出外显子56-60、71-74重复突变;患儿母亲P034探针(C)检出携带外显子61-70的重复突变,P035探针(D)检出携带外显子56-60、71-74的重复突变。 |
2.2 肌肉病理改变
病例1取左上肢肱二头肌行肌肉活检,病理改变以肌纤维萎缩、dystrophin蛋白缺失为特征(见图 3)。

|
图 3 病例1的肌肉活检结果 A(HE染色×10):肌纤维结构紊乱,大小不等,萎缩纤维及肥大纤维混杂分布,断面呈圆形、肌束膜、肌内膜纤维结缔组织增生(箭头所示);B(NADH-TR染色×20):极度萎缩肌纤维深染,部分肌纤维见虫蚀空洞(箭头所示);C(dystrophin免疫组织化学染色×40):dystrophin蛋白呈部分阳性表达,即分布于肌膜的棕褐色深染部分(箭头所示)。 |
2.3 脐血间充质干细胞移植后的血清酶谱变化
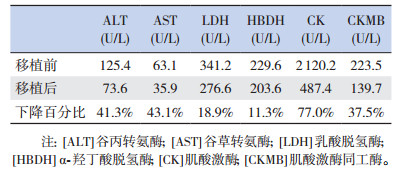
病例5行脐血间充值干细胞移植,移植5 d后ALT、AST、LDH、HBDH、CK、CKMB较移植前下降,以CK下降最显著(约下降77.0%),见表 2。
| 表 2 脐血间充质干细胞移植前后血清酶谱的变化 |
3 讨论
DMD是儿童期最常见的肌营养不良性疾病,发病率为1/4 000活产男婴[1]。但DMD起病隐匿,婴幼儿期多无症状,常导致诊断困难。2009年,英国的一项针对156例无明确家族史DMD患儿的回顾性分析发现,首发症状引起重视、首次去医疗机构就诊、首次检测肌酸激酶、通过基因检测或肌活检确诊的平均年龄分别为2.5岁、3.6岁、4.7岁、4.9岁,诊断平均延迟2.5年[2]。因此,提高对DMD的警惕,认识肌酸激酶检测的重要性并进一步行基因检测,是早期诊断的关键。
DMD因肌纤维缺乏dystrophin而发病,临床以进行性肌无力、肌萎缩为特点,受累患儿主要在儿童早期出现骨骼肌受累的表现。走路延迟可能是DMD患儿肌无力的最早表现,约60%的DMD患儿开始走路的平均年龄为17个月(13~23个月)[3]。但走路延迟及姿态的异常往往不易引起家长或医务工作者的重视,通过实验室检查发现CK、ALT、AST等升高方引起重视[4],而ALT、AST异常容易被误诊为病毒性肝炎或肝功能损害[5-6]。本研究6例患儿中1例(16.7%)初诊考虑肝功能损害,2例(33.3%)初诊考虑呼吸道感染,3例(50.0%)考虑肌病,均为血清酶谱尤其是CK显著升高才考虑到DMD的可能。CK是含量最丰富的肌肉特异性酶,共有5种同工酶,血清中检测到的主要为肌型,约占CK总量的95%[7]。CK > 正常上限1.5倍称为CK血症[8],反映肌肉受损,最常见于DMD[9]。据文献报道,患儿一旦发现CK血症,DMD可在随后的1.6个月(0~4个月)内确诊[3]。因此对于DMD疑似病例,应尽早行血清酶谱检测,如果显著升高,尤其是CK明显升高,应尽早行基因诊断。
DMD确诊依赖于基因检测或肌活检,检测到DMD致病基因Dystrophin突变可确诊;肌活检仅在基因检测不能确诊的情况下推荐,肌组织中dystrophin蛋白缺失可确诊[10-11]。Dystrophin基因是人类最大的基因之一,定位于Xp21.2,全长约2.5 Mb,编码79个外显子,突变频率高,突变形式多样[12]。突变类型中,缺失突变占55%~65%,存在中央区(外显子45-53)和5’端区(外显子2-20)两处缺失热区;重复突变占5%~15%;点突变占35%[13]。本研究6例患儿通过基因检测确诊,5例(83.3%)为缺失突变,1例(16.7%)为重复突变;其中1例患儿行肌活检,病理改变符合DMD肌营养不良。由于外显子缺失及重复突变共占比约80%,因此DMD基因诊断首先针对外显子缺失和重复突变进行;目前多重连接探针扩增法(MLPA)是检测Dystrophin基因外显子拷贝数异常、确诊DMD的首选方法[14]。MLPA采用通用引物,覆盖Dystrophin基因全部79个外显子,操作简便,具有高特异性和高通量特点。一旦发现患儿CK血症,应及时进行DMD基因突变的MLPA检测。DMD为X连锁隐性遗传病,因此应同时进行患儿母亲或姐妹的DMD基因检测,以检出家族中的女性携带者,便于提供遗传咨询和产前诊断。基因确诊的6例患儿中,2例患儿的母亲进行了DMD基因的MLPA检测,均检出携带致病基因。但是MLPA仅能检出缺失或重复突变(约占总突变的80%),因此对于MLPA检测阴性者,应采取其他技术(如PCR、测序)对其他类型的突变进行检测。
DMD起病隐匿,进展快,预后不良,目前尚无有效治疗措施。DMD的治疗主要包括:(1)激素治疗:目前唯一真正有效的治疗[15];(2)干细胞移植:利用干细胞多向分化的能力参与肌肉修复,目前已进入Ⅰ/Ⅱ期临床试验阶段[16];(3)基因治疗:通过腺病毒转染迷你或微小Dystrophin基因至异常的肌肉细胞以弥补肌营养不良蛋白表达的不足[17];(4)上调Utrophin蛋白表达:用Utrophin蛋白来代替dystrophin蛋白表达的不足[18];(5)终止密码子突变抑制疗法:抑制提前发生的终止密码子,以便dystrophin蛋白继续表达,有效的药物主要有氨基糖苷类抗生素和PCT124[19-20];(6)反义寡核苷酸外显子跳跃疗法:与前提mRNA互补结合以跳过突变的外显子序列,从而翻译成有功能的dystrophin蛋白[21]。本研究病例6行脐血间充质干细胞移植,移植5 d后的血清酶谱尤其CK较移植前明显下降,但是远期疗效还需长期随访。另外,本研究6例患儿的运动能力尚存,血清酶谱均表现为ALT、AST、LDH、HBDH、CK、CKMB同步升高,其中病例3在运动能力尚存的5年半随访期间6项酶谱呈波浪式进展,尤以CK的变化落差最显著,上述结果提示:(1)CK是反映骨骼肌损伤的敏感指标,DMD患儿的肌肉萎缩可能分批出现;(2)对于血清ALT、AST、CK同步升高者,推测DMD患儿体内尚存有功能的、正常的肌纤维[4],此期干预可能有效;(3)对于仅CK升高而ALT、AST、LDH、HBDH、CKMB均正常者,推测患儿体内已无功能性肌纤维[22],此期已丧失治疗时机。因此,血清酶谱的监测对于DMD患儿进行早期干预有指导意义。有研究提出,DMD患儿早期治疗(4岁前)可保护尚未病变的肌纤维、保持肌力,延缓运动功能的丧失和降低死亡率[23]。
综上所述,对于血清肌酶异常、运动功能异常或反复呼吸道感染的男童,应高度警惕DMD,尽早行基因检测以早期确诊。虽然目前DMD尚无有效治疗措施,但早期诊断有利于早期干预,以保护尚未病变的肌纤维,延缓疾病进展;而且家族中女性携带者的检出有利于遗传咨询和产前诊断。
| [1] | Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy[J]. Ann Neurol, 2012, 71 (3): 304–313. DOI:10.1002/ana.23528 |
| [2] | Ciafaloni E, Fox DJ, Pandya S, et al. Delayed diagnosis in Duchenne muscular dystrophy:data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet)[J]. J Pediatr, 2009, 155 (3): 380–385. DOI:10.1016/j.jpeds.2009.02.007 |
| [3] | van Ruiten HJ, Straub V, Bushby K, et al. Improving recognition of Duchenne muscular dystrophy:a retrospective case note review[J]. Arch Dis Child, 2014, 99 (12): 1074–1077. DOI:10.1136/archdischild-2014-306366 |
| [4] | McMillan HJ, Gregas M, Darras BT, et al. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy[J]. Pediatrics, 2011, 127 (1): e132–136. DOI:10.1542/peds.2010-0929 |
| [5] | 李素华, 陈益平, 陈均亚, 等. 进行性肌营养不良误诊为慢性肝炎5例[J]. 实用医学杂志, 2007, 23 (1): 126. |
| [6] | 刘平, 吴惧, 胡文广, 等. 儿童进行性肌营养不良误诊为病毒性肝炎五例临床分析[J]. 临床误诊误治, 2012, 25 (7): 40–42. |
| [7] | Brancaccio P, Lippi G, Maffulli N. Biochemical markers of muscular damage[J]. Clin Chem Lab Med, 2010, 48 (6): 757–767. |
| [8] | Kyriakides T, Angelini C, Schaefer J, et al. EFNS guidelines on the diagnostic approach to pauci-or asymptomatic hyperCKemia[J]. Eur J Neurol, 2010, 17 (6): 767–773. DOI:10.1111/j.1468-1331.2010.03012.x |
| [9] | 毛冰, 熊晖, 焦辉, 等. 肌酶分析在儿童肌病性高肌酸激酶血症鉴别诊断中的意义[J]. 北京大学学报 (医学版), 2014, 46 (1): 130–137. |
| [10] | Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1:diagnosis, and pharmacological and psychosocial management[J]. Lancet Neurol, 2010, 9 (1): 77–93. DOI:10.1016/S1474-4422(09)70271-6 |
| [11] | Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2:implementation of multidisciplinary care[J]. Lancet Neurol, 2010, 9 (2): 177–189. DOI:10.1016/S1474-4422(09)70272-8 |
| [12] | Blake DJ, Weir A, Newey SE, et al. Function and genetics of dystrophin and dystrophin-related proteins in muscle[J]. Physiol Rev, 2002, 82 (2): 291–329. DOI:10.1152/physrev.00028.2001 |
| [13] | Takeshima Y, Yagi M, Okizuka Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center[J]. J Hum Genet, 2010, 55 (6): 379–388. DOI:10.1038/jhg.2010.49 |
| [14] | 张媛媛, 刘晓亮, 何蓉, 等. 多重连接依赖探针扩增在假肥大型肌营养不良症家系基因诊断中的应用[J]. 中华医学遗传学杂志, 2014, 31 (3): 338–343. |
| [15] | GoemansN, Buyse G. Current treatment and management of dystrophinopathies[J]. Curr Treat Options Neurol, 2014, 16 (5): 287. DOI:10.1007/s11940-014-0287-4 |
| [16] | Bonfanti C, Rossi G, Tedesco FS, et al. PW1/Peg3 expression regulates key properties that determine mesoangioblast stem cell competence[J]. Nat Commun, 2015, 6 : 6364. DOI:10.1038/ncomms7364 |
| [17] | Seto JT, Bengtsson NE, Chamberlain JS. Therapy of genetic disorders-novel therapies for Duchenne muscular dystrophy[J]. Curr Pediatr Rep, 2014, 2 (2): 102–112. DOI:10.1007/s40124-014-0044-x |
| [18] | Guiraud S, Squire SE, Edwards B, et al. Second-generation compound for the modulation of utrophin in the therapy of DMD[J]. Hum Mol Genet, 2015, 24 (15): 4212–4224. DOI:10.1093/hmg/ddv154 |
| [19] | Scully MA, Pandya S, Moxley RT. Review of Phase Ⅱ and Phase Ⅲ clinical trials for Duchenne muscular dystrophy[J]. Expert Opinion Orphan Drugs, 2013, 1 (1): 33–46. DOI:10.1080/21678707.2013.746939 |
| [20] | Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy[J]. Muscle Nerve, 2014, 50 (4): 477–487. DOI:10.1002/mus.v50.4 |
| [21] | Nakamura A. X-linked dilated cardiomyopathy:A cardiospecific phenotype of dystrophinnopathy[J]. Pharmaceuticals, 2015, 8 (2): 303–320. DOI:10.3390/ph8020303 |
| [22] | Flanigan KM. Duchenne and Becker muscular dystrophies[J]. Neurol Clin, 2014, 32 (3): 671–688. DOI:10.1016/j.ncl.2014.05.002 |
| [23] | Merlini L, Gennari M, Malaspina E, et al. Early corticosteroid treatment in 4 Duchenne muscular dystrophy patients:14-year follow-up[J]. Muscle Nerve, 2012, 45 (6): 796–802. DOI:10.1002/mus.23272 |