 2017, Vol. 19
2017, Vol. 19



在体内,叶酸主要参与氨基酸的相互转换、DNA合成、甲基化反应等多种重要的代谢过程,与细胞增殖、分化密切相关[1]。研究表明,孕期叶酸缺乏可引起子代多种先天畸形的发生,如唇腭裂、神经管畸形等,孕期叶酸补充可降低神经管畸形的发生率,但并不能完全阻止神经管畸形的发生,提示可能有其他的机制参与。近年来随着表观遗传学研究的深入,越来越多的证据表明,孕期叶酸缺乏可通过改变子代基因组DNA甲基化模式而对其生长发育造成影响[2-3]。DNA甲基化是表观遗传学的重要组成部分,在维持基因组稳定,调控胚胎正常发育等方面起着重要作用[4]。胰岛素样生长因子(insulin-like growth factors, IGFs)是一类与细胞增殖、分化、凋亡等功能密切相关的生长调控因子,由具有特定功能的配体(IGF-1、IGF-2)、受体(IGF-1R、IGF-2R)及胰岛素样生长因子结合蛋白(IGFBPs)组成[5]。IGFs是胚胎和生后生长的重要调控因子,在生长发育中起着至关重要的作用。迄今,关于叶酸缺乏与IGFs间的关系研究很少,本实验通过构建叶酸缺乏的孕鼠模型,观察围孕期叶酸缺乏对胎鼠生长发育的影响,通过甲基化免疫共沉淀高通量测序技术(MeDIP-seq),探索叶酸缺乏组和正常对照组胎鼠的IGFs相关基因的DNA甲基化差异,同时检测孕鼠血清和胎鼠脑、肝脏组织中的IGF-1、IGFBP-3因子水平,初步探讨IGFs表达变化在孕鼠叶酸缺乏所致子代生长发育异常中的作用。
1 材料与方法 1.1 试剂与材料健康清洁级未经产成年Sprague-Dawley(SD)雌鼠22只及其饲料均购自成都达硕生物科技有限公司。叶酸、大鼠IGF-1、大鼠IGFBP-3酶联免疫吸附测定试剂盒均购自武汉伊莱瑞特生物科技有限公司。
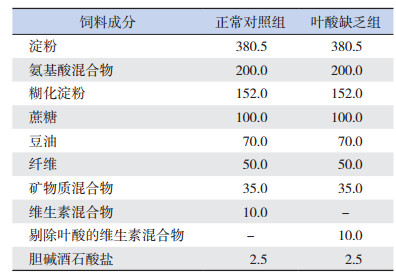
1.2 动物模型的制备将22只SD雌鼠随机分为叶酸缺乏组(n=12)和正常对照组(n=10)。叶酸缺乏组大鼠喂养饲料是在正常对照组基础上剔除叶酸,两组饲料配方详见表 1[5]。两组大鼠均自由饮水和进食,在喂养2周后与未行饮食干预的正常成年SD雄鼠1:1合笼交配。每日下午18:00合笼,次日上午7:00~8:00抽取雌鼠阴道分泌物于载玻片上,显微镜下观察到精子的当日计为孕第0天,随即转入普通饲养笼中,继续喂饲各组相应饲料直至取材。
| 表 1 两组大鼠饲料的组成成分(g/kg) |
1.3 标本采集及实验记录
两组雌鼠均于孕第20天称重后,腹腔注射10%水合氯醛(3.33 mL/kg)麻醉,剖腹取胎;然后快速剪开胸腔,暴露心脏,孕鼠心脏穿刺取血,室温放置2 h,2 230 r/min离心20 min,血清分装保存供后续ELISA分析用。每一窝胎鼠随机取6只迅速分离其脑、肝脏组织,PBS冲洗,液氮速冻,-80℃冰箱保存备用。每一窝胎鼠再随机取4只测量记录其头尾长、体重,并记录孕鼠每窝总活胎数。
1.4 ELISA法检测叶酸、IGF-1及IGFBP-3水平每一窝胎鼠随机取1只,即每组各取8只胎鼠脑及肝脏组织,采用ELISA法测定孕鼠血清IGF-1、IGFBP-3含量及胎鼠脑、肝脏组织中叶酸、IGF-1、IGFBP-3水平。具体操作步骤按照试剂盒说明书进行。
1.5 MeDIP-seq行全基因组DNA甲基化测序各组随机取3只非同窝胎鼠脑及肝脏组织行全基因组甲基化测序。使用组织基因组提取试剂盒(Omega公司,美国)提取胎鼠脑、肝脏组织DNA,DNA经Qubit Fluorometer定量仪和0.8%琼脂糖凝胶电泳质检合格后备用。将基因组DNA进行超声随机片段化,片段化后进行末端修复、3' 末端加A碱基,连接测序接头。用5-mC抗体对已连接测序接头的DNA进行沉淀,富集甲基化DNA片段,并对抗体富集结果进行qPCR验证,最后建立文库,文库检测合格后上机测序。
1.6 差异甲基化基因筛选根据MeDIP-seq测序结果,计算差异甲基化基因的差异倍数,利用统计学分析检验P值,若差异倍数 > 2,或≤1/2且P值 < 0.05,则表明两组间基因的甲基化存在差异。
1.7 统计学分析采用SPSS 20.0统计软件对数据进行统计学分析。正态分布的计量资料以均数±标准差(x± s)表示,两组间的比较采用两独立样本t检验,P < 0.05为差异有统计学意义。
2 结果 2.1 两组雌鼠受孕情况及胚胎情况对照组雌鼠10只,受孕8只;叶酸缺乏组雌鼠12只,受孕8只。对照组平均每窝胎鼠为15.0±1.3只,叶酸缺乏组平均每窝胎鼠为14.0±1.3只,差异无统计学意义(t=1.53,P > 0.05)。胎鼠的外观未见畸形,无脑膨出、脊柱裂等发育畸形。
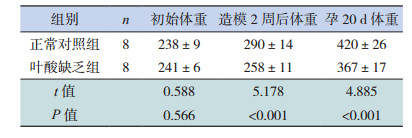
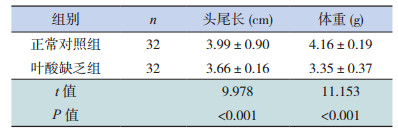
2.2 两组孕鼠及胎鼠体格生长指标两组孕鼠的初始体重比较差异无统计学意义(P > 0.05),但予以相应饲料2周后及孕第20天时,叶酸缺乏组孕鼠的体重远远低于正常对照组(P < 0.05)(表 2)。与正常对照组相比,叶酸缺乏组胎鼠平均头尾长、体重都显著降低(P < 0.05)(表 3)。
| 表 2 两组孕鼠体重的比较(x± s,g) |
| 表 3 两组胎鼠头尾长、体重的比较(x± s) |
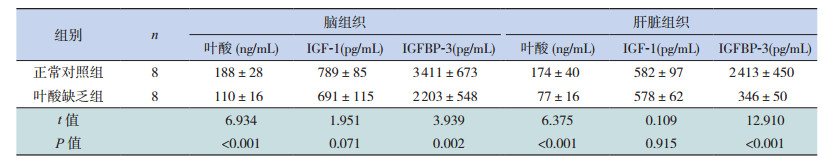
2.3 两组孕鼠血清IGF-1、IGFBP-3水平及两组胎鼠脑、肝脏组织中叶酸、IGF-1、IGFBP-3水平的比较
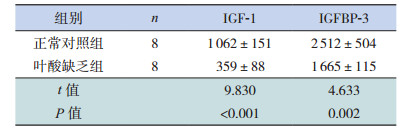
叶酸缺乏组孕鼠血清IGF-1、IGFBP-3水平及胎鼠脑、肝脏组织中叶酸、IGFBP-3水平均低于正常对照组(P < 0.05)。与正常对照组相比,叶酸缺乏组胎鼠脑、肝脏组织中IGF-1表达水平略下降,但差异无统计学意义(P > 0.05),见表 4~5。
| 表 4 两组孕鼠血清IGF-1和IGFBP-3水平比较(x± s,pg/mL) |
| 表 5 两组胎鼠脑及肝脏组织中叶酸、IGF-1、IGFBP-3水平比较(x± s) |
2.4 两组胎鼠脑、肝脏组织中IGFs基因的甲基化水平比较
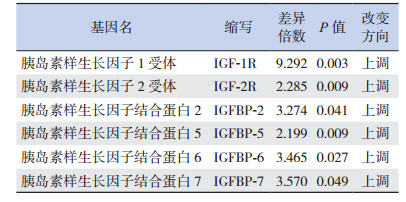
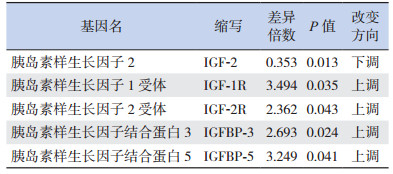
胎鼠脑组织IGFs基因的MeDIP测序结果显示:叶酸缺乏组IGF-1R、IGF-2R、IGFBP-2、IGFBP-5、IGFBP-6、IGFBP-7的甲基化水平均高于正常对照组(表 6);而在肝脏组织中,相比于正常对照组,叶酸缺乏组IGF-1R、IGF-2R、IGFBP-3、IGFBP-5甲基化水平增加,IGF-2甲基化水平则下降(表 7)。
| 表 6 与正常对照组比较叶酸缺乏组胎鼠脑组织甲基化水平变化(n=3) |
| 表 7 与正常对照组比较叶酸缺乏组胎鼠肝脏组织甲基化水平变化(n=3) |
3 讨论
在本研究中,我们发现与正常对照组相比,叶酸缺乏组胎鼠脑、肝脏组织叶酸浓度降低,且雌鼠体重,胎鼠头尾长、体重下降,但胎鼠无神经管畸形的发生。这与先前的多数研究结果[6-7]相一致。提示单纯的叶酸缺乏可能是导致胎鼠宫内生长发育迟缓的原因之一;而围孕期叶酸的缺乏则尚不足以引起胎鼠神经管畸形的发生,这可能与机体复杂的代偿调控机制密切相关。Kim等[8]构建了雄鼠叶酸缺乏的动物模型,发现雄鼠叶酸缺乏会降低胎鼠肝脏组织叶酸浓度,但脑组织叶酸浓度组间无差异。这与本研究结果并不一致,可能是因为相较于雄鼠的低叶酸,孕鼠的叶酸缺乏影响更为直接和严重,引起胎鼠包括脑组织在内的重要器官叶酸水平的下降。
本研究结果显示叶酸缺乏组孕鼠血清IGF-1、IGFBP-3浓度要远低于正常对照组,表明母体的营养会影响循环中IGF-1、IGFBP-3的水平。Bowman等[9]的研究表明循环中IGF-1减少,可降低组织器官对营养素的摄取能力,并使细胞增殖、分化、成熟障碍,可致使宫内生长迟缓的发生。研究发现宫内生长迟缓组孕后期母亲的IGF-1水平要明显低于正常对照组[10]。而另一项研究则发现与早产适于胎龄儿相比,早产宫内生长迟缓儿IGF-1、IGFBP-3浓度降低[11]。上述研究结果提示IGF-1、IGFBP-3参与了胎儿宫内生长受限的调控。本研究发现叶酸缺乏组胎鼠脑、肝脏组织的IGFBP-3水平都显著低于正常对照组,但IGF-1的水平在两组胎鼠脑、肝脏组织中则无明显差异。血循环中的IGFBP-3主要与IGF-1结合,其作用是延长IGF-1的半衰期,并可通过调控IGF-1对其受体的结合,进而促进或抑制IGF-1的生物学活性。此外,IGFBP-3也具有不依赖于IGF-1、直接调控细胞生长的作用[12]。出现上述现象的原因可能与IGFBP-3水平的降低有关。循环中IGFBP-3水平的降低,会降低其与IGF-1的结合率,使IGF-1作用半衰期缩短,清除率增加,则进一步降低循环中IGF-1的浓度,但却可增加IGF-1的组织利用率,从而使组织中的IGF-1下降不显著。
MeDIP-Seq的结果显示叶酸缺乏组胎鼠肝脏组织IGFBP-3的甲基化水平增加,且与脑组织相比,胎鼠肝脏组织的IGFBP-3下降更为显著。这可能是因为肝脏是IGFBP-3的最主要的合成部位,因此在肝脏组织差异最为明显。研究表明,特定基因的甲基化水平上调可导致基因沉默,而甲基化水平上调则可促进基因表达[1]。因此,基于本研究结果,我们推测孕鼠叶酸缺乏可能通过影响IGFBP-3的甲基化水平,进而导致基因表达的改变,可能参与胚胎生长发育的调控。
IGF-1主要调控出生后的生长发育,IGF-2则在胚胎期的生长调控中发挥作用。IGF-1R可介导IGF-1和IGF-2的许多生物学效应,但IGF-2R仅能与IGF-2结合。IGF-1R与配体结合后,可促进细胞有丝分裂,抑制细胞凋亡[13]。本研究通过对两组胎鼠IGFs相关基因的甲基化水平比较,发现与正常对照组相比,叶酸缺乏组脑、肝脏组织的IGF-1R、IGF-2R甲基化水平都增加,但IGF-2基因的甲基化水平则在肝脏组织中表现为下调,而IGF-1的甲基化水平则在组间无差异。以上结果提示了在胚胎发育期,主要是IGF-2参与了胎鼠生长发育的调控,而IGF-1R、IGF-2R甲基化的表达异常则可能导致相应受体表达的异常,引起相应细胞信号通路的异常而影响细胞增殖、分化,从而影响胚胎正常的生长发育。
IGFBPs不仅可调控IGF配体的生物活性,还可独立参与机体的多种生物学功能。研究表明[14],基因敲除IGFBP-2可使16周龄雄鼠的松质骨骨密度下降40%,而外源性给予IGFBP-2,则可促进成骨细胞的增殖。通过构建IGFBP-5基因过表达转基因小鼠,研究发现IGFBP-5基因的过表达会增加新生小鼠的死亡率,抑制宫内及青春期前的生长,损害肌肉的发育,并使雌鼠的生育能力下降[15]。而IGFBP-6基因过表达时,转基因小鼠出生后体重明显下降,小脑的大小和重量也降低。但基因敲除IGFBP-5或IGFBP-6,小鼠的表型则无明显变化,可能是通过对其他IGFBPs的调控来弥补上述基因的功能[16]。IGFBP-7则在调控细胞增殖分化、调控变态反应与参与机体免疫应答等方面有着重要的作用[17]。上述研究表明IGFBP-2、IGFBP-5、IGFBP-6、IGFBP-7在调控生长发育中发挥着重要的作用,而本研究发现叶酸缺乏组胎鼠脑组织IGFBP-2、IGFBP-5、IGFBP-6、IGFBP-7的甲基化水平均高于正常对照组,表明孕鼠叶酸缺乏会影响胎鼠IGFBPs的甲基化表达,可能通过干预上述基因的表达,而影响胎鼠的生长发育。
总之,本研究发现围孕期叶酸缺乏会导致胎鼠IGFs基因甲基化水平的改变,且伴随着孕鼠血清IGF-1、IGFBP-3的降低及胎鼠头尾长、体重的下降,提示围孕期叶酸缺乏可能通过影响IGFs基因的甲基化而对子代的生长发育造成影响。但本研究仅初步探讨了叶酸缺乏对子代DNA甲基化的影响,在后续研究中,将进一步验证IGFs基因的甲基化程度与相应基因表达的关系,深入探讨IGFs基因在叶酸缺乏所致胚胎发育异常中的机制。
| [1] | Ly A, Hoyt L, Crowell J, et al. Folate and DNA methylation[J]. Antioxid Redox Signal, 2012, 17 (2): 302–326. DOI:10.1089/ars.2012.4554 |
| [2] | Crider KS, Yang TP, Berry RJ, et al. Folate and DNA methylation:a review of molecular mechanisms and the evidence for folate's role[J]. Adv Nutr, 2012, 3 (1): 21–38. DOI:10.3945/an.111.000992 |
| [3] | Imbard A, Benoist JF, Blom HJ. Neural tube defects, folic acid and methylation[J]. Int J Environ Res Public Health, 2013, 10 (9): 4352–4389. DOI:10.3390/ijerph10094352 |
| [4] | McKay JA, Mathers JC. Diet induced epigenetic changes and their implications for health[J]. Acta Physiol (Oxf), 2011, 202 (2): 103–118. DOI:10.1111/apha.2011.202.issue-2 |
| [5] | Agrogiannis GD, Sifakis S, Patsouris ES, et al. Insulin-like growth factors in embryonic and fetal growth and skeletal development (Review)[J]. Mol Med Rep, 2014, 10 (2): 579–584. |
| [6] | 李屾, 梁良, 姚志刚, 等. 叶酸缺乏致胎鼠宫内发育迟缓及肝脏胰岛素生长因子系统表达变化[J]. 现代生物医学进展, 2014, 14 (4): 601–607. |
| [7] | Burgoon JM, Selhub J, Nadeau M, et al. Investigation of the effects of folate deficiency on embryonic development through the establishment of a folate deficient mouse model[J]. Teratology, 2002, 65 (5): 219–227. DOI:10.1002/(ISSN)1096-9926 |
| [8] | Kim HW, Kim KN, Choi YJ, et al. Effects of paternal folate deficiency on the expression of insulin-like growth factor-2 and global DNA methylation in the fetal brain[J]. Mol Nutr Food Res, 2013, 57 (4): 671–676. DOI:10.1002/mnfr.v57.4 |
| [9] | Bowman CJ, Streck RD, Chapin RE. Maternal-placental insulinlike growth factor (IGF) signaling and its importance to normal embryo-fetal development[J]. Birth Defects Res B Dev Reprod Toxicol, 2010, 89 (4): 339–349. DOI:10.1002/bdrb.20249 |
| [10] | Ferrero S, Mazarico E, Valls C, et al. Relationship between foetal growth restriction and maternal nutrition status measured by dual-energy X-ray absorptiometry, leptin, and insulin-like growth factor[J]. Gynecol Obstet Invest, 2015, 80 (1): 54–59. DOI:10.1159/000371761 |
| [11] | Chiesa C, Osborn JF, Haass C, et al. Ghrelin, leptin, IGF-1, IGFBP-3, and insulin concentrations at birth:is there a relationship with fetal growth and neonatal anthropometry?[J]. Clin Chem, 2008, 54 (3): 550–558. DOI:10.1373/clinchem.2007.095299 |
| [12] | Ranke MB. Insulin-like growth factor binding-protein-3(IGFBP-3)[J]. Best Pract Res Clin Endocrinol Metab, 2015, 29 (5): 701–711. DOI:10.1016/j.beem.2015.06.003 |
| [13] | Savage MO. Insulin-like growth factors, nutrition and growth[J]. World Rev Nutr Diet, 2013, 106 : 52–59. DOI:10.1159/000342577 |
| [14] | Clemmons DR. Role of IGF binding proteins in regulating metabolism[J]. Trends Endocrinol Metab, 2016, 27 (6): 375–391. DOI:10.1016/j.tem.2016.03.019 |
| [15] | Salih DA, Tripathi G, Holding C, et al. Insulin-like growth factor-binding protein 5(Igfbp5) compromises survival, growth, muscle development, and fertility in mice[J]. Proc Natl Acad Sci U S A, 2004, 101 (12): 4314–4319. DOI:10.1073/pnas.0400230101 |
| [16] | Bach LA. Insulin-like growth factor binding proteins 4-6[J]. Best Pract Res Clin Endocrinol Metab, 2015, 29 (5): 713–722. DOI:10.1016/j.beem.2015.06.002 |
| [17] | Akiel M, Rajasekaran D, Gredler R, et al. Emerging role of insulin-like growth factor-binding protein 7 in hepatocellular carcinoma[J]. J Hepatocell Carcinoma, 2014, 1 : 9–19. |