 2017, Vol. 19
2017, Vol. 19



先天性心脏病(congenital heart disease, CHD)发病率为6‰~8‰,居出生缺陷首位,是我国婴儿死亡的首要原因,严重危害我国儿童身心健康。因此,CHD发病机制的阐明对于其预防及早期干预具有重要的临床意义。心脏发育是一个极其复杂的过程,“心肌化”过程是保证胚胎心脏正常发育的关键环节。平面细胞极性(planar cell polarity, PCP)途径参与心肌细胞的黏附、运动和极化,通过精确调控心肌细胞定向迁移保证“心肌化”过程顺利完成。其中,Vangl2、Scrib及Rac1是PCP途径的关键分子,相关文献提示其表达缺失使心肌细胞迁移受到抑制,导致心脏流出道、心室肌发育异常[1]。
研究表明,40%~80% CHD的发生并非由于DNA序列的改变,而与环境-遗传交互作用有关。不良环境暴露可通过表观遗传学修饰使特定基因沉默或过表达,导致蛋白表型改变,最终引起疾病的发生。其中,组蛋白乙酰化/去乙酰化水平可通过改变染色质结构的疏密程度,调控转录因子与DNA结合,决定靶基因表型。该动态平衡如受干扰,将使心肌细胞相关基因表达异常,导致CHD发生[2],组蛋白去乙酰化酶(histone deacetylase, HDAC)是参与这一调控的关键酶,其表达水平异常与组蛋白乙酰化/去乙酰化失衡密切相关[2]。然而,组蛋白乙酰化/去乙酰化失衡是否可引起心肌细胞PCP关键分子的变化迄今尚未阐明。丙戊酸(valproic acid, VPA)为短链脂肪酸类HDAC抑制剂,可抑制HDAC表达及活性,导致组蛋白乙酰化水平升高,进而干扰组蛋白乙酰化/去乙酰化的动态平衡,且本团队前期研究提示VPA可抑制C2C12肌细胞肌相关蛋白表达下调,抑制其成肌分化[3]。因此,本研究利用VPA干扰H9C2心肌细胞乙酰化/去乙酰化修饰的动态平衡,评估HDAC1~3及Vangl2、Scrib、Rac1表达水平的变化,探讨组蛋白乙酰化/去乙酰化失衡时PCP关键分子的变化,从表观遗传学角度认识心脏早期发育的调控通路。
1 材料与方法 1.1 实验动物及细胞来源6~8周健康成年清洁级野生型C57/B6小鼠,体重20~25 g,购自四川大学动物实验中心,H9C2心肌细胞购自中国科学院武汉生物细胞库。
1.2 主要仪器及试剂胎牛血清、MEM/F12培养基(美国Corning公司);VPA、蛋白酶抑制剂Cocktail(美国Sigma-Aldrich公司);RIPA裂解液(强)(北京索莱宝科技有限公司);TRIzol(美国Life Technology公司);RNA逆转录试剂盒(日本Takara公司);SsoFast EvaGreen PCR试剂盒(美国Bio-Rad公司);HDAC1~3及Vangl2一抗(美国Proteintech公司);Scrib、Rac1一抗(美国Santa Cruz公司);GAPDH(北京康为世纪生物科技有限公司);HDAC活性试剂盒(美国BioVision公司)。酶标仪(美国Thermo Scientific公司);徕卡倒置相差显微镜DM2000(德国Leica微系统有限公司);荧光定量PCR仪(美国Bio-Rad公司)。
1.3 H9C2心肌细胞培养常规复苏H9C2心肌细胞,使用含有10%胎牛血清、100 U/mL青霉素及100 μg/mL链霉素的MEM/F12培养基在37℃、5%CO2饱和湿度下培养于T75培养瓶中,每隔2~3 d传代1次,当细胞数量达到实验所需时,将细胞用0.25%胰酶(美国GIBCO公司)消化后1 200 r/min离心3 min,弃去上清液,加入MEM/F12培养基重悬细胞,调整细胞含量为2×105/mL,每孔2 mL移入6孔板中,待细胞密度达70%~80%开始干预。
1.4 实验分组将小鼠于25℃环境下饲养,自由饮食、摄水,相对湿度50%~60%,噪音80分贝以下,通风换气15~20次/h。随机选取发情期雌鼠与雄鼠以3:1合笼,查见阴栓者记为孕0.5 d(E0.5 d)。
本研究团队前期预实验结果显示[4]:孕鼠能够耐受VPA的最大剂量为700 mg/kg,此时流产率约30%~50%,超过这一剂量,孕鼠流产率高达80%~100%,因此本研究选择最大剂量VPA 700 mg/kg处理孕鼠;小鼠胚胎心脏发育的关键时期为E11.5~E14.5 d,故选择于E10.5 d对孕鼠进行干预。随机分配上述健康清洁级野生型C57/B6孕鼠为空白对照组(n=10)、溶剂对照组(n=10)和VPA干预组(n=20)。VPA干预组孕鼠于E10.5 d腹腔注射VPA 700 mg/kg(溶剂为生理盐水);溶剂对照组孕鼠腹腔注射等量生理盐水;空白对照组不做任何处理。
参考相关文献,VPA干预细胞的浓度范围为1.0~10.0 mmol/L[5-7],结合本研究团队前期研究[3],故选用2.0、4.0及8.0 mmol/L浓度梯度VPA对细胞进行干预。将细胞分为空白对照组、溶剂对照组及VPA干预组。空白对照组不进行任何处理,溶剂对照组加入等量生理盐水,VPA干预组予不同浓度梯度VPA处理细胞。所有样品同时置于5%CO2的37℃培养箱中培养,按照细胞所要求的培养基种类每天更换培养基并进行相同的处理。分别于处理后24、48及72 h采集细胞行下一步检测。
1.5 胚胎心脏苏木精-伊红染色于E10.5 d断颈处死孕鼠,打开腹腔、分离子宫,取出胚胎后于解剖显微镜下分离胎心。PBS清洗后于10%福尔马林固定2 h。60%、70%、80%、95%及100%梯度酒精脱水各1 h。二甲苯透明后石蜡包埋、心脏连续切片(主动脉弓顶部-心尖),行苏木精-伊红(HE)染色,观察胚胎心脏结构发育异常。
1.6 RNA提取及cDNA合成每组平行设3个复孔,TRIzol裂解细胞,严格按照试剂盒说明书提取总RNA,采用Nanodrop_2000分光光度计对提取后的RNA进行浓度及纯度检测。采用RNA逆转录试剂盒严格按照说明书操作,将1 μg RNA逆转录为cDNA。
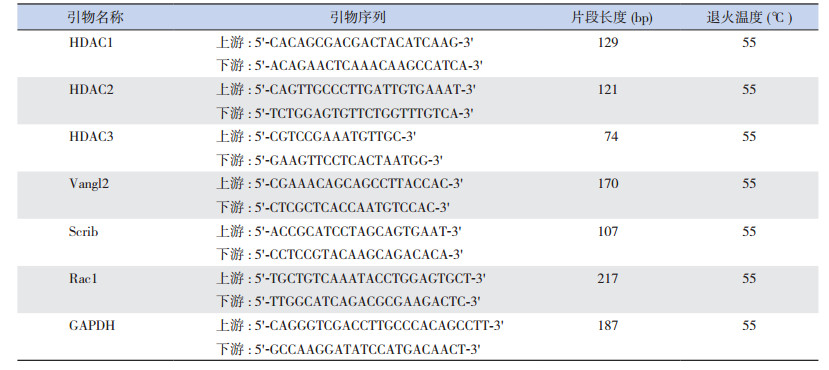
1.7 实时荧光定量PCR本研究所有引物均经过扩增效率检测,扩增效率均 > 95%。使用SsoFast EvaGreen PCR试剂盒进行qRT-PCR。体系总量为10 μL:EvaGreen 5 μL,无核酸酶水3 μL,正反向引物各0.5 μL,cDNA 1 μL。PCR扩增条件为:95℃ 30 s;95℃ 5 s,55℃ 10 s,共39个循环;而后65~95℃绘制熔解曲线,基因相对表达量以2-△△Ct表示。各基因引物序列、片段长度及退火温度见表 1。实验独立重复3次。
| 表 1 PCR引物序列、长度及退火温度 |
1.8 Western blot检测
每组平行设3个复孔,用预冷PBS清洗细胞数次,使用RIPA(强)与蛋白酶抑制剂(1:25混合)裂解细胞,冰上作用20 min,4℃9 661 r/min离心15 min,留取上清液,BCA法测定样品蛋白浓度。蛋白变性后,采用8%十二烷基硫酸钠聚丙烯酰胺凝胶电泳分离胶(SDS-polyacrylamide gel, SDS-PAGE),每孔加入50 μg总蛋白电泳,通过100 V电转2 h将蛋白印迹至PVDF膜上,5%脱脂奶(TBST配置)室温封闭1 h,加入封闭液稀释的一抗HDAC1(1:1 000)、HDAC2(1:1 000)、HDAC3(1:500)、Vangl2(1:1 000)、Scrib(1:500)、Rac1(1:200)、GAPDH(1:500),置于4℃冰箱孵育过夜。TBST洗膜10 min×3次,加入相应二抗室温孵育90 min,TBST洗膜10 min×3次后,采用化学发光法进行显色,最后采用Bio-Rad成像系统采集图像,利用Gelpro32软件进行半定量分析。目的蛋白相对表达量采用其灰度值与相应内参(GAPDH)灰度值之比表示。实验独立重复3次。
1.9 总HDAC活性检测利用比色法测定总HDAC活性,具体实验步骤参照HDAC酶活性检测试剂盒说明书:(1)裂解细胞提取蛋白并测定蛋白浓度,在圆底96孔酶标板中每孔加入100 μg蛋白,每组平行设3个复孔,并加入无核酸酶水稀释至85 μL;加入75 μL无核酸酶水稀释10 μL Hela细胞核提取液作为阳性对照;83 μL无核酸酶水中加入2 μL曲古抑素A作为阴性对照。(2)每孔加10 μL 10×HDAC分析缓冲液及5 μL HDAC发光底物充分混合,37℃孵育1 h。(3)每孔加入10 μL赖氨酸显色剂,充分混匀,37℃孵育30 min。(4)利用酶标仪测定A405 nm处的光密度值,根据标准曲线计算出每个样品的总HDAC活性[μmol/(L · μg)]。实验独立重复3次。
1.10 统计学分析采用SPSS 16.0统计学软件对数据进行统计学分析,计量资料以均数±标准差(x± s)表示,多样本均数比较采用方差分析,采用Tukey法进行组间多重比较;计数资料以百分率(%)表示,多组间比较采用χ2检验;P < 0.05为差异有统计学意义。计数资料组间两两比较采用卡方分割法,调整P < 0.017为差异有统计学意义。
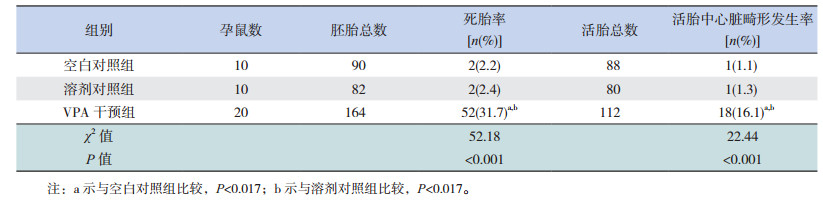
2 结果 2.1 VPA干预孕鼠致胎鼠心脏畸形的发生情况VPA干预组胎鼠死亡率较空白及溶剂对照组显著增加(P < 0.05);胎鼠心脏HE染色观察心脏结构发育异常显示,上述3组活胎鼠中心脏畸形发生率分别为16.1%、1.1%、1.3%,VPA干预组心脏畸形发生率显著高于两对照组(P < 0.05)(表 2);VPA干预组心脏畸形主要表现为室间膈缺损(VSD)(14/18)、心室壁变薄伴心肌致密化不全(NVM)(4/18)(图 1),而空白对照组和溶剂对照组均为1例房间隔缺损(ASD)。
| 表 2 VPA干预孕鼠致胎鼠心脏畸形发生率的比较 |

|
图 1 VPA致胎鼠心脏发育异常(苏木精-伊红染色) [LA]左心房;RA[右心房];[LV]左心室;[RV]右心室;[IVS]室间隔;[VSD]室间隔缺损;[AO]主动脉;[PA]肺动脉;[TV]三尖瓣;[MV]二尖瓣。对照组室间隔发育完整,心室壁致密、增厚;VPA干预组室间隔见VSD(箭头所示);与空白及溶剂对照组相比,VPA干预组心腔明显变小,并见心室壁菲薄伴心肌致密化不全(方框所示)。 |
2.2 VPA对H9C2细胞HDACs mRNA及蛋白表达水平的影响
与空白及溶剂对照组比较,HDAC1于VPA 2.0、4.0及8.0 mmol/L干预后24、48、72 h mRNA表达水平均明显上调(P < 0.05);而与mRNA变化相反,蛋白表达水平于干预后48 h及72 h显著降低(P < 0.05)。与空白及溶剂对照组比较,HDAC2于VPA 2.0、4.0及8.0 mmol/L干预后,仅在24 h时mRNA表达显著下降(P < 0.05),蛋白表达水平于干预后各时间点均显著下降(P < 0.05)。与空白及溶剂对照组比较,HDAC3 mRNA于VPA 4.0及8.0 mmol/L干预后各时间点表达均明显上调(P < 0.001),而蛋白表达水平于VPA 2.0、4.0及8.0 mmol/L干预后各时间点均显著降低(P < 0.01)。不同浓度VPA干预组间,于各时间点分别比较差异均无统计学意义(P > 0.05)。见图 2。

|
图 2 各组HDACs mRNA及蛋白表达水平比较(n=3) A~C:VPA干预对HDAC1 mRNA及蛋白水平的影响;D~F:VPA干预对HDAC2 mRNA及蛋白水平的影响;G~I:VPA干预对HDAC3 mRNA及蛋白水平的影响。a示与空白及溶剂对照组比较,P < 0.05。 |
2.3 VPA对H9C2细胞PCP关键分子mRNA及蛋白表达水平的影响
与空白及溶剂对照组相比,Vangl2及Scrib mRNA表达水平于不同浓度VPA干预后48 h及72 h均显著下降(P < 0.05);Vangl2蛋白表达水平于干预后72 h显著下降(P < 0.05);Scrib蛋白表达水平于干预后48 h及72 h均显著下调(P < 0.05)。Rac1 mRNA及蛋白表达水平与空白及溶剂对照组比较,差异均无统计学意义(P > 0.05)。不同浓度VPA干预组间,于各时间点分别比较差异均无统计学意义(P > 0.05)。见图 3。

|
图 3 各组PCP关键分子mRNA及蛋白表达水平比较(n=3) A~C:VPA干预对Vangl2 mRNA及蛋白水平的影响;D~F:VPA干预对Scrib mRNA及蛋白水平的影响;G~I:VPA干预对Rac1 mRNA及蛋白水平的影响。a示与空白及溶剂对照组比较,P < 0.05。 |
2.4 VPA对H9C2细胞总HDAC活性的影响
与空白及溶剂对照组比较,VPA 4.0、8.0 mmol/L干预后24 h,总HDAC活性显著降低(P < 0.05),而2.0 mmol/L VPA干预组较空白及溶剂对照组总HDAC活性无显著变化(P > 0.05);干预后48 h及72 h,3个剂量VPA干预组总HDAC活性均显著低于空白及溶剂对照组(P < 0.05)。不同浓度VPA干预组间,于各时间点分别比较差异均无统计学意义(P > 0.05)。见图 4。

|
图 4 各组总HDAC活性比较(n=3) a示与空白及溶剂对照组比较,P < 0.05。 |
3 讨论
胚胎心脏正常发育需要众多基因在不同时间和空间顺次精确表达,如果这些关键基因表达异常,将不同程度干扰胚胎心脏的分子调控程序化进程,最终导致CHD的发生。PCP途径中的Vangl2、Scrib及Rac1是与心脏早期发育密切相关的3种关键蛋白,其表达缺失将干扰“心肌化”过程,引起心脏发育异常:Vangl2参与心室肌与流出道的发育,基因突变致胎鼠心肌细胞分化异常,不能伸出板状和丝状伪足,细胞极化、迁移受抑制[8];Scrib与心肌细胞间黏附有关,表达缺失可致心脏袢化异常、VSD、心室壁变薄及心肌致密化不全[9];Rac1缺失可导致心肌细胞变为球形、心室小梁化异常、细胞凋亡增加[10]。因此,进一步研究PCP关键分子的表达及调控,成为CHD病因探讨中的关键问题。
近年研究显示,表观遗传学修饰调控着不同环境下基因的表达模式,是外界环境改变致疾病发生、发展的关键机制。表观遗传学修饰所致的组蛋白乙酰化状态改变是环境因素致畸的重要原因[11-12]。其中,HDAC是参与去乙酰化表观遗传学调控的关键酶:可去除组蛋白赖氨酸残基的乙酰化基团,使染色质结构致密,阻碍转录因子与DNA结合,抑制转录[13]。其中,Ⅰ类HDAC中的HDAC1~3组织分布最为广泛,参与心肌细胞分化、脂代谢,在心脏早期发育过程中起着至关重要的作用,其表达缺失可导致心肌细胞过度增生、造成扩张性心肌病、右心室肥厚与闭塞等多种先天性心脏发育异常[14-16]。因此,HDAC作为一种重要的表观遗传调控因子,对转录具有“开-关”作用;而PCP通路在“心肌化”过程中对心肌细胞极化、迁移起重要调控作用,阐明两者之间的调控作用将有利于更深层次地从表观遗传学角度探寻环境因素所致CHD的分子生物学机制。
VPA作为一种传统的抗惊厥药物被广泛应用于临床[17]。同时,它也是一种HDAC抑制剂,主要可通过抑制Ⅰ类HDAC活性,发挥抗增殖、促凋亡等抗肿瘤作用[13]。但是,VPA具有潜在致畸作用,妊娠期使用可致神经管、心血管、骨骼等多种畸形[18]。本研究证实,VPA可致胎鼠心脏结构发育异常,主要表现为VSD、心室壁变薄及心肌致密化不全,这些病理改变与PCP关键分子表达缺失所致病理改变有诸多相似之处,推测VPA可能通过干扰PCP途径导致胚胎心脏发育异常。因此,本研究在细胞水平,利用VPA干预大鼠胚胎心肌细胞H9C2,进一步探讨其对HDAC及PCP相关基因的影响。研究结果显示,VPA可上调HDAC1、3 mRNA表达水平,随VPA浓度增加,表达水平逐渐升高,而两者蛋白表达变化与mRNA变化恰好相反,mRNA表达上调与蛋白表达下调相平行,且均在干预后48 h及72 h最为显著;HDAC2 mRNA及蛋白表达水平变化一致,均为下降,于干预后24 h最为明显,提示VPA干预对HDAC1、3的作用机制有别于HDAC2。国外相关文献亦报道,VPA对不同HDAC的作用具有选择性、差异性及细胞特异性:Bradbury等[19]报道VPA可通过增加H3K4甲基化水平,选择性上调HL60白血病细胞株中HDAC11 mRNA及蛋白表达水平,而对人宫颈癌细胞株HDAC11表达没有影响;Kwiecińska等[5]研究结果亦显示,VPA可选择性抑制人卵巢癌细胞HDAC2、7蛋白表达水平,而对其他HDAC无影响;Krämer等[20]利用VPA干预鼠畸胎瘤细胞和人肾胚胎瘤细胞发现,VPA可选择性抑制HDAC2蛋白表达,而其mRNA表达水平没有变化,进一步实验证实VPA可通过泛素化酶E2激活蛋白酶体加速HDAC2蛋白降解而降低其蛋白表达水平。结合相关文献报道,我们推测,本研究中VPA干预后HDAC1、3 mRNA表达升高伴随蛋白表达下降的其中一种可能解释为:VPA可选择性作用于HDAC1、3,参与转录后调控,使其蛋白错误折叠、蛋白构象发生改变,进而被从内质网清除,由泛素-蛋白酶体系统降解[21-22],而蛋白水平下降导致mRNA代偿性升高[23]。总HDAC活性测定证实了VPA对HDAC活性的抑制作用,且也于干预后48 h及72 h最为显著,与HDAC1~3的蛋白表达水平降低时间点一致,表明HDAC1~3是H9C2中最主要的去乙酰化酶,也提示其表达被抑制可能是导致乙酰化/去乙酰化失衡的重要原因。为明确VPA对PCP关键分子的影响,本实验对Vanlg2、Scrib及Rac1的表达水平进行检测,结果显示Vanlg2、Scrib mRNA及蛋白表达均被抑制,mRNA表达下降于干预后48 h最为明显,而蛋白表达则于72 h最为显著,mRNA与蛋白变化的时序不一致性可能与蛋白的表达峰度及半衰期相关[24-25]。
上述结果显示,HDAC抑制剂VPA可抑制HDAC1~3蛋白表达及功能,HDAC下调同时Vangl2、Scrib mRNA及蛋白表达水平亦下调,提示HDAC1~3可能对Vangl2、Scrib存在正向调控作用,而非组蛋白去乙酰化修饰发挥的负向调控作用。既往文献表明:基因敲除小鼠胚胎干细胞中HDAC1表达后采用基因微阵列技术分析发现,约7%的基因表达受影响,其中2/3基因表达上调(HDAC1为靶基因的抑制因子),而余1/3表达下调(提示HDAC1为靶基因的激活因子),提示HDAC对基因的调控作用远不仅局限于组蛋白去乙酰化修饰[26]。除组蛋白外,HDAC也可对非组蛋白进行翻译后修饰,发挥正向调控作用。这种激活作用既可位于转录水平,即HDAC作为转录激活因子(如对Gja1的调控)或与其他转录因子共同形成复合物(如对糖皮质激素受体的调控)直接参与转录激活,也可位于转录后水平[14, 26-27]。
因此,本研究结果证实VPA可直接抑制HDAC1~3蛋白表达水平及总HDAC活性,导致乙酰化/去乙酰化失衡,同时下调PCP途径关键分子Vanlg2、Scrib mRNA及蛋白表达水平,这可能是环境因素致CHD发生的机制之一。但是,VPA对Ⅰ类HDAC的作用机制以及HDAC是否与Vangl2、Scrib相结合共同参与胚胎心肌细胞“心肌化”过程尚待进一步明确。目前,多种HDAC调控药物已应用于临床[13],一旦两者之间的调控网络被阐明,将对CHD的一级预防及早期干预产生一定的临床实用价值。
| [1] | Davis EE, Katsanis N. Cell polarization defects in early heart development[J]. Circ Res, 2007, 101 (2): 122–124. DOI:10.1161/CIRCRESAHA.107.157446 |
| [2] | Gallinari P, Di Marco S, Jones P, et al. HDACs, histone deacetylation and gene transcription:from molecular biology to cancer therapeutics[J]. Cell Res, 2007, 17 (3): 195–211. |
| [3] | 李一飞, 华益民, 方婕, 等. 组蛋白乙酰化/去乙酰化失衡对C2C12肌原细胞成肌分化的影响[J]. 中华妇幼临床医学杂志, 2014, 10 (5): 36–41. |
| [4] | Wu G, Nan C, Rollo JC, et al. Sodium valproate-induced congenital cardiac abnormalities in mice are associated with the inhibition of histone deacetylase[J]. J Biomed Sci, 2010, 17 : 16. DOI:10.1186/1423-0127-17-16 |
| [5] | Kwiecińska P, Wróbel A, Taubøll E, et al. Valproic acid, but not levetiracetam, selectively decreases HDAC7 and HDAC2 expression in human ovarian cancer cells[J]. Toxicol Lett, 2014, 224 (2): 225–232. DOI:10.1016/j.toxlet.2013.10.035 |
| [6] | Barbetti V, Gozzini A, Cheloni G, et al. Time-and residuespecific differences in histone acetylation induced by VPA and SAHA in AML1/ETO-positive leukemia cells[J]. Epigenetics, 2013, 8 (2): 210–219. DOI:10.4161/epi.23538 |
| [7] | Fedier A, Dedes KJ, Imesch P, et al. The histone deacetylase inhibitors suberoylanilide hydroxamic (Vorinostat) and valproic acid induce irreversible and MDR1-independent resistance in human colon cancer cells[J]. Int J Oncol, 2007, 31 (3): 633–641. |
| [8] | Ramsbottom SA, Sharma V, Rhee HJ, et al. Vangl2-regulated polarisation of second heart field-derived cells is required for outflow tract lengthening during cardiac development[J]. PLoS Genet, 2014, 10 (12): e1004871. DOI:10.1371/journal.pgen.1004871 |
| [9] | Phillips HM, Rhee HJ, Murdoch JN, et al. Disruption of planar cell polarity signaling results in congenital heart defects and cardiomyopathy attributable to early cardiomyocyte disorganization[J]. Circ Res, 2007, 101 (2): 137–145. DOI:10.1161/CIRCRESAHA.106.142406 |
| [10] | Leung C, Lu X, Liu M, et al. Rac1 signaling is critical to cardiomyocyte polarity and embryonic heart development[J]. J Am Heart Assoc, 2014, 3 (5): e001271. DOI:10.1161/JAHA.114.001271 |
| [11] | Sadowski SL. Congenital cardiac disease in the newborn infant:past, present, and future[J]. Crit Care Nurs Clin North Am, 2009, 21 (1): 37–48. DOI:10.1016/j.ccell.2008.10.001 |
| [12] | Huhta J, Linask KK. Environmental origins of congenital heart disease:The heart-placenta connection[J]. Semin Fetal Neonatal Med, 2013, 18 (5): 245–250. DOI:10.1016/j.siny.2013.05.003 |
| [13] | Abend A, Kehat I. Histone deacetylases as therapeutic targets-from cancer to cardiac disease[J]. Pharmacol Ther, 2015, 147 : 55–62. DOI:10.1016/j.pharmthera.2014.11.003 |
| [14] | Eom GH, Kook H. Posttranslational modifications of histone deacetylases:implications for cardiovascular diseases[J]. Pharmacol Ther, 2014, 143 (2): 168–180. DOI:10.1016/j.pharmthera.2014.02.012 |
| [15] | Kim YS, Kim MJ, Koo TH, et al. Histone deacetylase is required for the activation of Wnt/β-catenin signaling crucial for heart valve formation in zebrafish embryos[J]. Biochem Biophys Res Commun, 2012, 423 (1): 140–146. DOI:10.1016/j.bbrc.2012.05.098 |
| [16] | Reller MD, Strickland MJ, Riehle-Colarusso T, et al. Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005[J]. J Pediatr, 2008, 153 (6): 807–813. DOI:10.1016/j.jpeds.2008.05.059 |
| [17] | Peterson GM, Naunton M. Valproate:a simple chemical with so much to offer[J]. J Clin Pharm Ther, 2005, 30 (5): 417–421. DOI:10.1111/jcp.2005.30.issue-5 |
| [18] | Jentink J, Dolk H, Loane MA, et al. Intrauterine exposure to carbamazepine and specific congenital malformations:systematic review and case-control study[J]. BMJ, 2010, 341 : c6581. DOI:10.1136/bmj.c6581 |
| [19] | Bradbury CA, Khanim FL, Hayden R, et al. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors[J]. Leukemia, 2005, 19 (10): 1751–1759. DOI:10.1038/sj.leu.2403910 |
| [20] | Krämer OH, Zhu P, Ostendorff HP, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2[J]. EMBO J, 2003, 22 (13): 3411–3420. DOI:10.1093/emboj/cdg315 |
| [21] | Thapar R, Denmon AP. Signaling pathways that control mRNA turnover[J]. Cell Signal, 2013, 25 (8): 1699–1710. DOI:10.1016/j.cellsig.2013.03.026 |
| [22] | Wakabayashi-Nakao K, Tamura A, Furukawa T, et al. Quality control of human ABCG2 protein in the endoplasmic reticulum:ubiquitination and proteasomal degradation[J]. Adv Drug Deliv Rev, 2009, 61 (1): 66–72. DOI:10.1016/j.addr.2008.08.008 |
| [23] | Koraichi F, Videmann B, Mazallon M, et al. Zearalenone exposure modulates the expression of ABC transporters and nuclear receptors in pregnant rats and fetal liver[J]. Toxicol Lett, 2012, 211 (3): 246–256. DOI:10.1016/j.toxlet.2012.04.001 |
| [24] | Chen G, Gharib TG, Huang CC, et al. Discordant protein and mRNA expression in lung adenocarcinomas[J]. Mol Cell Proteomics, 2002, 1 (4): 304–313. DOI:10.1074/mcp.M200008-MCP200 |
| [25] | Tian Q, Stepaniants SB, Mao M, et al. Integrated genomic and proteomic analyses of gene expression in Mammalian cells[J]. Mol Cell Proteomics, 2004, 3 (10): 960–969. DOI:10.1074/mcp.M400055-MCP200 |
| [26] | Zupkovitz G, Tischler J, Posch M, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1[J]. Mol Cell Biol, 2006, 26 (21): 7913–7928. DOI:10.1128/MCB.01220-06 |
| [27] | Cao Y, Lu L, Liu M, et al. Impact of epigenetics in the management of cardiovascular disease:a review[J]. Eur Rev Med Pharmacol Sci, 2014, 18 (20): 3097–3104. |