 2017, Vol. 19
2017, Vol. 19



2. 上海交通大学医学院附属上海儿童医学中心医学遗传科, 上海 200127;
3. 深圳市宝安区妇幼保健院儿科,广东 深圳 518000
Prader-Willi综合征(Prader-Willi syndrome, PWS, OMIM: 176270)是一种罕见的遗传性疾病,是由父源性染色体15q11.2-q13区域缺失所致的基因组印记病,临床特点包括肌张力低下、婴儿期喂养困难、多食、肥胖、性腺发育不良、成年身高受损、认知和行为障碍[1-2]。PWS发病率约1/10 000~1/30 000[3]。国内关于PWS散在报道,多围绕其临床特征与遗传学诊断,关于PWS患者尤其是PWS女性患者内分泌代谢特点的研究较少。PWS是症状性病态肥胖最常见的原因之一,早期诊断和合理干预对改善患儿生活质量、预防严重并发症和延长寿命至关重要[3-4]。根据不同年龄患者的表型特点,针对内分泌代谢紊乱进行有效干预是十分必要的。本研究收集上海儿童医学中心2015年11月至2016年4月期间诊治的4例Prader-Willi综合征患者临床资料,分析其内分泌代谢特点。
1 资料与方法 1.1 研究对象4例PWS患者来自4个无血缘关系的家庭,均无类似疾病家族史。均根据2015年中国Prader-Willi综合征诊治专家共识[3]进行诊断。
病例1:女,10岁8个月,因精神运动发育落后10年就诊。曾因吸吮能力差、肌张力低多次住院治疗。4岁前喂养困难、嗜睡、少动、不哭,4岁后食量大、肥胖。患儿为第一胎第一产,3~4岁能独坐,5岁6个月能独走,至今上楼需手扶。4~5岁能有意识叫“爸爸、妈妈”,现就读小学2年级,能用词语、短句沟通,数数能从0~8,与同龄人交流少,不愿活动,易疲劳。睡眠打鼾。性格固执,脾气稍暴躁,喜抓挠皮肤。体查:身高116 cm( < P3),BMI 25.3 kg/m2( > P97),向心性肥胖,特殊面容(长脸、窄面、杏仁眼、小嘴)。小手、小脚,手背饱满,手指呈锥形、尺侧缘弧度消失。四肢可见抓痕。双乳未触及乳核,外阴色素沉着,无阴毛、腋毛。PWS临床评分9分。
病例2:女,11岁2个月,肥胖10年。出生体重3.5 kg,身长52 cm。生后混合喂养。1岁以后食欲增加,体态渐肥胖,限制饮食后肥胖改善不明显。患儿8个月因髋关节脱位行石膏固定;3岁行斜视矫正手术;10岁跌倒后右侧胫腓骨骨折,予手术内固定。患儿为第一胎第一产。1岁余能独走,1岁4个月能叫“爸爸、妈妈”,现能简单对答,发音较含糊。就读特殊学校,与同学相处愉快,可以写字、认字、辨认颜色,能数数及完成简单加减法。患儿性格固执。睡眠打鼾。月经来潮1年(3次月经),不规则。体查:身高153 cm(P50~P75),BMI 30.5 kg/m2( > P97),向心性肥胖,特殊面容(长脸、窄面、杏仁眼、小嘴,嘴角向下),无黑棘皮,皮肤紫纹以腰背部及腹部较集中。小手、小脚,手背饱满,手指呈锥形。双乳B2级,未触及乳核,外阴见阴毛生长、P3级。PWS评分8.5分。
病例3:女,11岁,因肥胖8年就诊。患儿第二胎第二产,剖宫产出生,无窒息抢救史,出生体重3.2 kg。生后人工喂养,3岁前喂养困难、少动、不哭;3岁后突然食量增加,体重增长加速,体态渐肥胖。患儿7个月能坐,1岁5个月能扶走,2岁能独走。1岁余能叫“爸爸”,现能短句交流,发音不清楚。语言表达、计算能力显著落后于同龄儿,就读于特殊学校,学习困难。脾气暴躁,固执,喜挠皮肤。睡眠打鼾。体查:身高133 cm( < P3),BMI 37.8 kg/m2( > P97),向心性肥胖,颈部、双侧腋下见黑棘皮,特殊面容(长颅,窄面,杏仁眼,嘴角向下)。小手、小脚,手背饱满,手指呈锥形、尺侧缘弧度消失。双乳B3级,外阴少许阴毛,外阴P2级。PWS临床评分8.5分。
病例4:女,5岁10个月,体重增长过快3年。患儿2岁后突然食量增大,体重增长加速。患儿为第一胎第一产,出生体重3.5 kg,身长52 cm。生后人工喂养,12个月能翻身,20个月能坐、爬,21个月会扶站,2岁能独走。语言发育落后,现仍吐字不清。脾气暴躁,性格较固执。曾多次因“重症肺炎、呼吸功能不全”住院治疗。曾外院诊断“重度阻塞性呼吸暂停综合征、腺样体肥大”。体查:身高110 cm(P3~P10),BMI 28.1 kg/m2( > P97),向心性肥胖,颈部见黑棘皮,特殊面容(长颅,窄面,杏仁眼)。小手、小脚,手背饱满,手指呈锥形、尺侧缘弧度消失。双乳B2级,可及乳核,外阴P1级。PWS临床评分8分。
1.2 基因组DNA提取采集患者外周血2 mL,乙二胺四乙酸(EDTA)抗凝,充分混匀,取200 μL全血,使用德国Qiagen公司QIAamp DNA Blood Mini Kit试剂盒提取基因组DNA,NanoDrop 2000分光光度计测得DNA浓度及比值(OD 260/280比值1.8~2.0),DNA样本-20℃保存待用。
1.3 PWS的遗传学检测取5 μL(50 ng/μL)患儿DNA样本,并设2份阴性对照,采用荷兰MRC-Holland公司SALSA MS-MLPA Prader-Willi/Angelman(ME028-B2)试剂盒进行15q11.2-q13染色体致病区域拷贝变异及甲基化状态的检测。DNA变性、杂交、连接,PCR扩增,PCR产物纯化后使用ABI3730测序仪进行毛细管电泳,电泳结果采用GeneMarker®软件(V2.2.0版本,美国SoftGenetics公司)分析。
1.4 口服葡萄糖耐量试验试验采集患儿清晨空腹静脉血,查血糖、胰岛素、C肽、HbA1c、胰岛自身抗体等。空腹血糖、HbA1c正常则行口服葡萄糖耐量试验(oral galucose tolerance test, OGTT):口服葡萄糖1.75 g/kg(最大量75 g),30 min、60 min、120 min后查血糖、胰岛素、C肽。
胰岛素抵抗指数(IR)=空腹血糖(mmol/L)×空腹胰岛素(mIU/L)/22.5。IR > 2.69可判断为胰岛素抵抗。
1.5 戈那瑞林激发试验检测黄体生成素(LH)、卵泡刺激素(FSH)的基础值,并肌注戈那瑞林(LHRH)2.5 μg/kg、最大量100 μg,注射后30 min、60 min、90 min、120 min检测LH和FSH。若LH峰值≥5 U/L,LH峰值/FSH峰值≥0.6,考虑中枢性性腺轴启动。
2 结果 2.1 PWS的遗传学检测例1和例4存在15q11.2-q13区域父源性缺失;例3的15q11.2-q13区域拷贝数正常,但甲基化结果提示为母源性同源二倍体(即两条同源染色体均来自母亲,父源性染色体缺失);例2的15q11.2-q13区域拷贝数正常,甲基化状态未发现明显异常。
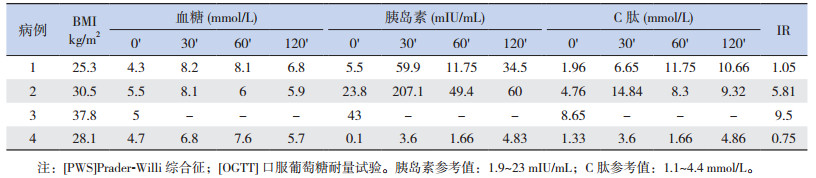
2.2 OGTT试验结果例3多次空腹血糖为9~10 mmol/L,餐后血糖最高达19 mmol/L,HbA1c 7.4%,糖尿病自身抗体:胰岛素抗体(AIA)、抗胰岛细胞抗体(CIA)和抗谷氨酸脱羧酶抗体(GADA)均阴性,诊断为2型糖尿病。给予控制饮食、二甲双胍治疗,血糖控制在正常范围,服药2月左右自行停用,仅予饮食控制,血糖控制较满意(餐前 < 7 mmol/L,餐后 < 11.1 mmol/L)。
其余3例空腹血糖、HbA1c正常,OGTT试验(见表 1)提示:例2存在胰岛素抵抗,例4胰岛素分泌无明显升高,3例患者的胰岛素分泌高峰在30 min和120 min。
| 表 1 4例PWS患者的OGTT试验结果 |
2.3 LHRH激发试验及乳腺、性腺B超结果
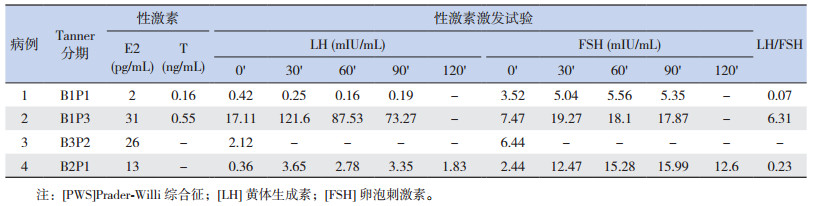
性激素及激发试验(表 2)提示例2为中枢性性腺轴启动;例1和例4不支持中枢性性腺轴启动。例3在青春期出现第二性征,血E2升高,B超示子宫卵巢增大,符合青春期改变,故未行LHRH激发试验。
| 表 2 4例PWS患者的性激素基础值及激发试验结果 |
例2 B超提示子宫、卵巢发育,乳腺未见腺体样组织。例1超声提示子宫、乳腺未发育。例4超声可见乳腺、子宫、卵巢增大。
2.4 其他资料例1和例4的血脂正常,例2甘油三脂升高,例3甘油三脂、胆固醇均升高。4例患者腹部B超均提示脂肪肝,其中例3出现肝功能异常,ALT最高达127 U/L,AST最高达155 U/L。例3超声提示皮下脂肪瘤。4例患儿的尿酸均无异常。例2、例4的25-羟维生素D3降低,分别为14.9 ng/mL和19.22 ng/mL。4例患儿的FT4、TSH均正常;B超提示例1甲状腺回声欠均匀、血流少,例4甲状腺质地不均、右叶囊性结节。4例患儿的血皮质醇和ACTH检测未见异常。
3 讨论PWS是一组特征性的临床症候群,诊断主要依靠临床评分与分子遗传诊断[3]。通过中文数据库检索至2016年11月,我国共报道约100例PWS病例,大部分患者在新生儿期或婴幼儿期确诊[5-8]。本研究的病例就诊时已出现不同程度的内分泌异常,均在学龄期或青春期确诊,其中3例患者通过对15q11.2-q13致病区域拷贝数变异及甲基化状态的检测确诊,1例未能找到遗传学依据。
PWS患者在儿童期可出现过度摄食、肥胖,常继发糖尿病、心肺功能不全、阻塞性睡眠呼吸暂停、血栓性静脉炎等肥胖相关并发症[4]。国外研究[9]表明,25%的PWS患者成年后出现糖尿病,PWS患者糖尿病的平均发病年龄是20岁,大部分为2型糖尿病,少数为1型糖尿病;儿童期发生糖尿病的机率很低,仅0~3.6%。本研究病例3于11岁诊断继发性2型糖尿病,可能与高BMI有关;另3例患者的胰岛素反应峰值在30 min和120 min,其中1例合并胰岛素抵抗。这种胰岛素的双峰分泌在既往PWS或肥胖儿童合并胰岛素抵抗的研究中未见报道[10-11],但成人2型糖尿病患者可有类似情况,可能为胰岛素分泌相对不足所致。本研究发现PWS患者还存在脂代谢异常,4例患儿的腹部B超均提示脂肪肝,其中1例肝酶显著升高;2例患儿的胆固醇和/或甘油三脂增高;1例B超发现皮下脂肪瘤。而PWS的报道对脂代谢关注较少,其脂代谢特点有待进一步研究。
PWS还是一种下丘脑功能障碍性疾病,约60%患儿的头颅MRI显示垂体发育不全,80%伴生长激素缺乏,49%伴性腺功能低下,30%伴中枢性甲状腺功能减退,部分伴有中枢性肾上腺皮质功能不全[2]。由于本研究的患儿性格异常、智力障碍,加之年龄较大、镇静剂效果欠佳,未能配合完成垂体MRI检查。患儿中2例身高低于同性别同年龄儿童身高的P3,1例为P3~P10,1例身高正常。有学者认为,PWS患儿在出现肥胖之前早期使用重组生长激素治疗,不仅可改善身高,更重要的是可降低体脂率,提高肌肉质量,调整血脂水平,获得更好的运动机能[12]。本文4例患者均有使用生长激素的指征,但均已出现严重肥胖、未控制的阻塞性睡眠呼吸暂停、未控制的糖尿病等生长激素治疗禁忌症,不适合生长激素治疗。
大部分PWS存在性腺功能低下,表现为外生殖器发育不良,青春期发育延迟,性发育变异,不孕不育等。可能与垂体-下丘脑-性腺轴功能低下有关。但2012年Siemensma等[13-14]对68例男性和61例女性PWS患者进行了性腺功能的纵向研究,认为PWS患者的性腺功能障碍可能为原发性。本研究4例女性患者就诊时均未发现明显外生殖器畸形,病例1第二性征延迟,病例3青春期有性发育启动,病例4性早熟。病例2性激素激发试验提示中枢性性发育启动,但乳腺发育与外周激素水平升高不一致,仍需进一步研究。由于PWS患者随时可能出现性发育停滞,应注意监测动态监测性腺发育情况,不建议合并性早熟的PWS患者使用促性腺激素释放激素类似物治疗。而女性PWS患者如在青春期有正常或接近正常的第二性征:乳房发育或月经来潮,性激素替代治疗可以促进其性生理正常化,并使其在育龄期获得生育机会。因此,例可考虑予性激素替代治疗,改善性腺功能。
部分PWS患者可合并中枢性甲状腺功能低下,国内有PWS合并桥本氏甲状腺炎的报道[15]。本研究4例患者的FT4、FSH均在正常范围,但2例B超发现甲状腺质地不均匀,需要定期监测甲状腺功能。少部分PWS患者可能合并肾上腺皮质功能不全。本研究4例患儿的血皮质醇和ACTH均无异常,无肾上腺皮质功能减退依据。10岁以后的PWS患者脊柱侧弯发生率可达30%~80%,可能与椎旁肌肌张力低,以及过度肥胖和严重骨质疏松有关[2, 16]。本研究4例患者均无脊柱侧弯,但其中2例在轻微外力撞击下出现骨折,可能与他们的血清25-羟维生素D3显著下降有关。
综上所述,PWS患者存在多种内分泌功能紊乱,有针对性地合理干预有利于提高患儿生活质量、改善预后。因此,需要注意PWS患者的随访及管理。
| [1] | Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome:a review of clinical, genetic, and endocrine findings[J]. J Endocrinol Invest, 2015, 38 (12): 1249–1263. DOI:10.1007/s40618-015-0312-9 |
| [2] | Emerick JE, Vogt KS. Endocrine manifestations and management of Prader-Willi syndrome[J]. Int J Pediatr Endocrinol, 2013, 2013 (1): 14. DOI:10.1186/1687-9856-2013-14 |
| [3] | 中华医学会儿科学分会内分泌遗传代谢学组. 中国Prader-Willi综合征诊治专家共识[J]. 中华儿科杂志, 2015, 53 (6): 419–424. |
| [4] | Elena G, Bruna C, Benedetta M, et al. Prader-Willi syndrome:clinical aspects[J]. J Obes, 2012, 2012 (10): 473941. |
| [5] | 詹实娜, 何玺玉, 王春枝, 等. 新生儿Prader-Willi综合征13例临床表型分析[J]. 中国循证儿科杂志, 2012, 7 (3): 200–204. |
| [6] | 刘虎, 钟家蓉. 新生儿Prader-Willi综合征5例临床报道[J]. 中外女性健康研究, 2015, 8 (16): 198–199. |
| [7] | 王萍, 周伟, 魏谋, 等. 新生儿Prader-Willi综合征7例并文献复习[J]. 中华实用儿科临床杂志, 2013, 28 (20): 1571–1574. DOI:10.3969/cma.j.issn.2095-428X.2013.20.015 |
| [8] | 黄新疆, 毛晓健, 刘丽, 等. 中国南方Prader-Willi综合征患儿27例临床表现及分子特征分析[J]. 中华实用儿科临床杂志, 2016, 31 (8): 573–578. |
| [9] | Diene G, Mimoun E, Feigerlova E, et al. Endocrine disorders in children with Prader-Willi syndrome-data from 142 children of the French database[J]. Horm Res Paediatr, 2010, 74 (2): 121–128. DOI:10.1159/000313377 |
| [10] | 黄永兰, 林文春, 黄俊菁. 肥胖儿童胰岛素抵抗和代谢综合征的临床研究[J]. 中国儿童保健杂志, 2011, 19 (2): 164–166. |
| [11] | 朱延华, 杨黛稚, 李津, 等. Prader-Willi综合征患者一例胰岛素敏感性分析及文献复习[J]. 中华糖尿病杂志, 2012, 4 (3): 159–161. |
| [12] | Deal CL, Tony M, Hoybye C, et al. Growth hormone research society workshop summary:consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome[J]. J Clin Endocrinol Metab, 2013, 98 (6): E1072–1087. DOI:10.1210/jc.2012-3888 |
| [13] | Siemensma EP, de Lind van Wijngaarden RF, Otten BJ, et al. Testicular failure in boys with Prader-Willi syndrome:longitudinal studies of reproductive hormones[J]. J Clin Endocrinol Metab, 2012, 97 (3): E452–E459. DOI:10.1210/jc.2011-1954 |
| [14] | Siemensma EP, van Alfen-van der Velden AA, Otten BJ, et al. Ovarian function and reproductive hormone levels in girls with Prader-Willi syndrome:a longitudinal study[J]. J Clin Endocrinol Metab, 2012, 97 (3): E1766–E1773. |
| [15] | 高力敏. 1例Prader-Willi综合征合并桥本氏甲状腺炎临床及分子遗传学研究[D]. 太原: 山西医科大学, 2011. |
| [16] | 许德荣, 李书纲. 1例Prader-Willi综合征合并脊柱侧凸患儿的诊治报道[J]. 中华骨与关节外科杂志, 2015, 8 (6): 531–533. |