 2017, Vol. 19
2017, Vol. 19

2. 重庆医科大学附属儿童医院核医学科,儿童发育疾病研究教育部重点实验室,儿童发育重大疾病国家国际科技合作基地,儿科学重庆市重点实验室,儿童肿瘤研究室,重庆 400014

 , ZHU Min, DENG Lei-Li, LEI Pei-Yun, LUO Yan-Hong, ZENG Yan, ZHU Gao-Hui, SONG Cui
, ZHU Min, DENG Lei-Li, LEI Pei-Yun, LUO Yan-Hong, ZENG Yan, ZHU Gao-Hui, SONG Cui
X-连锁低血磷性佝偻病(X-Linked hypophos-phatemic rickets, HYP或XLH, OMIM #307800)是低血磷性佝偻病中最常见的一种,发病率约为1/20 000[1-2]。XLH症状多于1岁以后症状明显,多为X-连锁显性遗传,发病机制为磷酸盐调节基因(phosphate regulating gene with homologies to endopeptidases on the X-chromosome, PHEX)突变导致血清成纤维生长因子23(FGF23)水平升高,影响肾小管对磷的重吸收,导致尿磷排泄增多、血磷水平下降[3-4]。XLH早期表现为骨骼和牙不同程度的损害,如方颅、鸡胸、漏斗胸、肋骨串珠、肋外翻、手足镯,出牙延迟、易脱落、牙痛,直立行走后出现下肢骨软化症状:“X”或“O”型腿,骨骼疼痛、生长迟缓等[5-6];后期可出现牙脓肿、关节活动障碍[6-7]。补充磷制剂可改善其临床症状。
国内外已报道444例PHEX基因突变所致的XLH患者,多发生于外显子(约占79%),包括缺失、插入、错义及无义突变等;部分发生于内含子(约占19%),包括剪接位点突变;极少数发生于非翻译区(untranslated regions, UTR)[8-9]。XLH的基因变异具有明显的异质性,多种突变形式可引起相似的临床表现。本研究采用PCR-Sanger测序方法,分析2例低血磷性佝偻病患儿的PHEX基因,发现2种未见报道的PHEX基因新突变。
1 资料与方法 1.1 研究对象先证者1,男,3岁4个月,发现“O型腿”2年。不伴骨痛。第一胎第一产、足月顺产出生,出生时身长、体重不详,出生时无窒息。母乳喂养,10月龄添加辅食,无偏食挑食。7月龄能独坐、10月龄独站、1岁2个月能独立行走。既往体健。患儿父母健康,否认近亲结婚,家族中无类似患者。体格检查:身高82 cm(-4.34 SD),体重11 kg,前额突出,无方颅,无鸡胸,无肋骨串珠,无郝氏沟,无手足镯,心肺腹部查体未见异常,“O型腿”、膝间距9 cm。实验室检查:血钙正常,血磷1.04 mmol/L(参考值:1.3~2.1 mmol/L),ALP 792.8 U/L(参考值:95~405 U/L),25(OH) D3正常,PTH正常。血气分析无异常。下肢骨片(见图 1):双膝关节骨骺端稍膨大,临时钙化带模糊。肾脏B超无异常。

|
图 1 先证者1的髋关节、下肢X片 股骨远端及近端钙化带模糊,如箭头所示。 |
先证者2,女,7岁4个月,发现“X型腿”伴双下肢间断性疼痛5年。第一胎第一产、足月顺产出生,出生身长、体重不详,出生时无产伤、无窒息。喂养史、生长发育史无特殊。既往体健。患儿父母体健,否认近亲结婚,家族中无类似患者。体格检查:身高111.2 cm(-2.61 SD),体重22.5 kg,无方颅,无鸡胸,无郝氏沟,无肋串珠,无手足镯,心肺腹部查体未见明显异常,“X型腿”、踝间距12 cm。实验室检查:血钙正常,血磷0.5 mmol/L(参考值:1.3~2.1 mmol/L),ALP 428 U/L(参考值:95~405 U/L),PTH 110 pg/mL(参考值:10~69 pg/mL)。肾脏B超:双肾皮质回声稍增强,双侧肾椎体见少许钙质成分沉着。
1.2 高通量捕获测序、突变验证及家系分析通过重庆医科大学附属儿童医院伦理审查及患儿家属知情同意后,采集患儿及其父母空腹EDTA抗凝静脉血各2 mL,提取基因组DNA。DNA浓度 > 10 ng/μL,体积 > 100 μL;纯度应达到A260/280 > 1.8,A260/230 > 1.2。
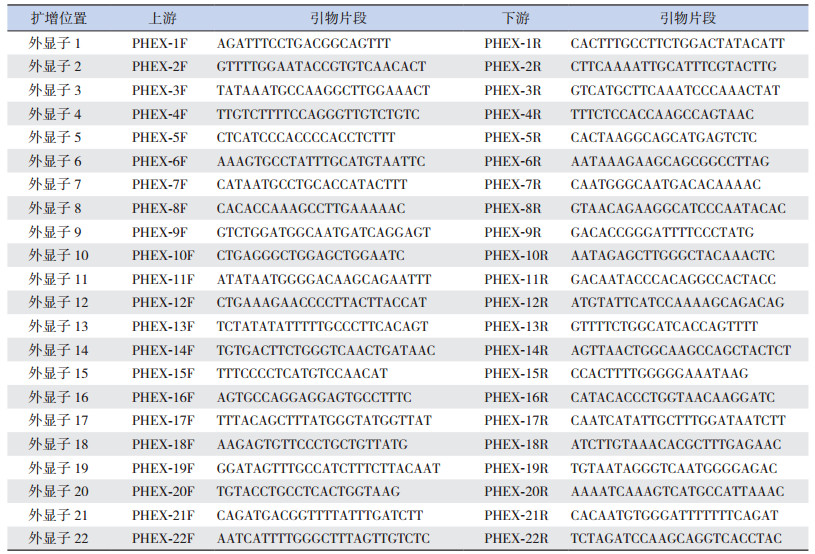
取先证者基因组,对PHEX基因的外显子区及两侧的剪接位点序列进行高通量测序,并利用PCR-Sanger测序对先证者及家系进行突变验证和分析(中国人民解放军空军医学研究所附属医院病理检验中心完成)。引物设计根据GeneBank人类PHEX基因序列(NG_007563.2),见表 1。PCR扩增,取PCR样品用ABI遗传分析仪进行测序。阳性突变者用另一份备用样品进行复测。
| 表 1 PHEX基因引物序列 |
2 结果 2.1 基因测序结果
经高通量捕获测序,发现先证者1在PHEX基因第14号内含子剪切位点发生单碱基置换:IVS14+1G > A;运用PCR-Sanger测序进行突变验证及家系分析,证实患儿父母该基因位点正常。见图 2。先证者2的PHEX基因检测到1个杂合突变:位于8号外显子的移码突变c.931dupC;运用PCR-Sanger测序进行突变验证及家系分析,发现患儿父母该基因位点正常。见图 3。

|
图 2 先证者1及家系的PHEX基因测序图 先证者1在第14号内含子发生单碱基置换:IVS14+1G > A,突变位点如箭头所示;其父母该位点正常。 |

|
图 3 先证者2及家系的PHEX基因测序 先证者2在第8号外显子发生碱基插入突变:c.931dupC,突变位点如箭头所示;其父母该位点正常。 |
经查询Locus Database数据库,PHEX基因的c.931dupC和IVS14+1G > A两个突变均未见报道。查阅PUBMED(https://www.ncbi.nlm.nih.gov/pubmed/)和CNKI(http://www.cnki.net/)及万方(http://www.wanfangdata.com.cn/)文献数据库,均无该基因突变的文献报道。因此,本文检测到的2种PHEX基因突变为新突变。经MUTATION TASTER(www.mutationtaster.org/)生物信息学分析,2例突变均为致病性突变,但未行相关基因功能学研究。
3 讨论至2016年11月,PHEX database数据库已报道444例XLH患者,共计338种PHEX基因突变,其中由中国人报道的突变共计13种。PHEX基因突变中移码突变约占27%,错义突变占21%,剪接位点突变占19%。但中国人群中最多见的PHEX基因突变类型是无义突变(约占30%),其次为错义突变(约占22%),剪接位点突变约占21%。本研究2例XLH患儿检测到PHEX基因突变,分别为移码突变c.931dupC和剪接位点突变IVS14+1G > A,均为国际上首次报道的新突变。在我国已报道的XLH病例中[9-14, 16-17],PHEX基因突变以E18、E22外显子突变更为多见;而在PHEX数据库中,以E09、E22外显子突变较为常见。这提示PHEX基因突变呈散在分布,可能与种族、性别、地区等因素相关。
本研究先证者1的IVS14+1G > A突变位于14号内含子,MUTATION TASTER(www.mutationtaster.org/)生物信息学分析提示突变“GA-A-GA”为经典的剪切位点突变方式,可影响外显子的剪切,导致外显子跳跃,产生不完整的氨基酸链,合成异常蛋白。PHEX数据库中共报道了4种类型的内含子14(I14)基因突变,其中3例为碱基缺失(IVS14,c.1584+3delGAGT;IVS14+1,c.1586+1delGA;IVS14+3,c.1586+3delGAGT),另1例为同区域剪接位点碱基突变(IVS14-1,c.1587-1G > C)导致剪接位点异常[8, 18-20]。2012年Beck-Nielsen等[8]报道1例31岁的女性低血磷性佝偻病患者在PHEX基因内含子14位置上发生突变:IVS14-1,c.1587-1G > C,引起内含子保留和外显子跳跃,患者身高157 cm(-1.8 SD),表现为轻微骨骼畸形,严重牙病变,无骨折史;血磷低,1, 25(OH)2D3水平正常,PTH轻度升高,骨密度正常。本研究先证者1发生的IVS14+1G > A突变,影响了外显子剪切,可能导致外显子15的跳跃而产生不完整的氨基酸链。本研究与Beck-Nielsen等报道的患者相比,血生化表现一致,但尚无牙病变,下肢畸形较严重,身高较正常同龄儿减低。这可能与种族、年龄或性别不同有关,也可能PHEX基因突变即使发生在同一内含子不同位置,疾病的表现在也可不一致。2013年,Yue等[14]发现3例患儿分别在内含子I10(c.1174-1G > A)、I15(c.1646-2A > T)、I17(c.1768+2T > G)发生突变,这3例患儿均为男性,起病年龄0.75~5岁,2例有家族史、1例为散发,均有“O型腿”表现,无牙龈病变,与本研究的患儿表现一致。这表明基因突变的类型与表型不一定有明显的相关性。
本研究先证者2检测到PHEX基因位于8号外显子的c.931dupC移码突变:p.Gln311Profs*13,MUTATION TASTER(www.mutationtaster.org/)生物信息学分析提示,它可导致311位的氨基酸由Gln(谷氨酰胺)转变为Pro(脯氨酸),在323位出现终止密码“UGA”,编码的氨基酸序列转变为P PVRLA GLHQE GH*,导致编码氨基酸提前终止、PHEX蛋白结构及功能异常,为致病性突变。该患儿骨骼畸形明显,提示移码突变产生的截短蛋白可能导致严重的表型。目前PHEX database数据库已报道6例外显子8突变(C.871C > T,c.931C > T,c.888+2G > T,c.904A > G,c.914T > C,c.897_898del),其中5例点突变(2例错义突变、3例无义突变),1例为缺失突变[1, 18, 20-23]。目前PHEX数据库中尚无8号外显子插入突变的报道。
本研究2例XLH患儿检测到2个国际上首次报道的PHEX基因新突变,丰富了XLH的致病基因谱,有助于深入认识PHEX基因的异质性。
| [1] | Francis F, Strom TM, Hennig S, et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets[J]. Genome Res, 1997, 7 (6): 573–585. DOI:10.1101/gr.7.6.573 |
| [2] | Bowe AE, Finnegan R, Jan de Beur SM, et al. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate[J]. Biochem Biophys Res Commun, 2001, 284 (4): 977–981. DOI:10.1006/bbrc.2001.5084 |
| [3] | Xia WB, Jiang Y, Li M, et al. Levels and dynamic changes of serum fibroblast growth factor 23 in hypophosphatemic rickets/osteomalacia[J]. Chin Med J (Eng1), 2010, 123 (9): 1158–1162. |
| [4] | Liu S, Zhou J, Tang W, et al. Pathogenic role of Fgf23 in Hyp mice[J]. Am J Physiol Endocrinol Metab, 2006, 291 (1): E38–E49. DOI:10.1152/ajpendo.00008.2006 |
| [5] | Thakker RV, O'Riordan JL. Inherited forms of rickets and osteomalacia[J]. Baillieres Clin Endocrinol Metab, 1988, 2 (1): 157–191. DOI:10.1016/S0950-351X(88)80012-0 |
| [6] | Clausmeyer S, Hesse V, Clemens PC, et al. Mutational analysis of the PHEX gene:novel point mutations and detection of large deletions by MLPA in patients with X-linked hypophosphatemic rickets[J]. Calcif Tissue Int, 2009, 85 (3): 211–220. DOI:10.1007/s00223-009-9260-8 |
| [7] | Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia:a search for gender, race, anticipation, or parent of origin effects on disease expression in children[J]. J Clin Endocrinol Metab, 1996, 81 (11): 4075–4080. |
| [8] | Beck-Nielsen SS, Brixen K, Gram J, et al. Mutational analysis of PHEX, FGF23, DMP1, SLC34a3 and CLCN5 in patients with hypophosphatemic rickets[J]. J Hum Genet, 2012, 57 (7): 453–458. DOI:10.1038/jhg.2012.56 |
| [9] | 宋莹, 麻宏伟, 黎芳, 等. X-连锁低磷性佝偻病的基因突变分析[J]. 中国当代儿科杂志, 2013, 15 (11): 928–931. DOI:10.7499/j.issn.1008-8830.2013.11.002 |
| [10] | 许莉军, 夏维波. 低血磷性佝偻病的分子遗传学研究[D]. 北京: 北京协和医学院, 2015. |
| [11] | 王静, 金春莲. 抗维生素D佝偻病PHEX基因诊断研究[D]. 沈阳: 中国医科大学, 2007. |
| [12] | 唐佳, 潘敬新, 蒋玮莹, 等. 一例抗维生素D佝偻病的基因诊断和新突变的致病性鉴定[J]. 中华临床医师杂志, 2012, 6 (5): 1226–1230. |
| [13] | Yuan L, Wu S, Xu H, et al. Identification of a novel PHEX mutation in a Chinese family with X-linked hypophosphatemic rickets using exome sequencing[J]. Biol Chem, 2015, 396 (1): 27–33. |
| [14] | Yue H, Yu JB, He JW, et al. Identification of two novel mutations in the PHEX gene in Chinese patients with hypophosphatemic rickets/osteomalacia[J]. PLoS One, 2013, 9 (5): e97830. |
| [15] | Holm IA, Nelson AE, Robinson BG, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets[J]. J Clin Endocrinol Metab, 2001, 86 (8): 3889–3899. DOI:10.1210/jcem.86.8.7761 |
| [16] | Kang QL, Xu J, Zhang Z, et al. Three novel PHEX gene mutations in four Chinese families with X-linked dominant hypophosphatemic rickets[J]. Biochem Biophys Res Commun, 2012, 423 (4): 793–798. DOI:10.1016/j.bbrc.2012.06.042 |
| [17] | Zhu X, Li M, Pan H, et al. Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in 24 sporadic patients with Rett syndrome in China[J]. J Child Neurol, 2010, 25 (7): 842–848. DOI:10.1177/0883073809350722 |
| [18] | Gaucher C, Walrant-Debray O, Nguyen TM, et al. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets[J]. Hum Genet, 2009, 125 (4): 401–411. DOI:10.1007/s00439-009-0631-z |
| [19] | Filisetti D, Ostermann G, von Bredow M, et al. Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues[J]. Eur J Hum Genet, 1999, 7 (5): 615–619. DOI:10.1038/sj.ejhg.5200341 |
| [20] | Holm IA, Huang X, Kunkel LM, et al. Mutational analysis of the PHEX gene in patients with X-linked hypophosphatemic rickets[J]. Am J Hum Genet, 1997, 60 (4): 790–797. |
| [21] | Sato K, Tajima T, Nakae J, et al. Three novel PHEX gene mutations in Japanese patients with X-linked hypophosphatemic rickets[J]. Pediatr Res, 2000, 48 (4): 536–540. DOI:10.1203/00006450-200010000-00019 |
| [22] | Rowe PS, Oudet CL, Francis F, et al. Distribution of mutations in the PEX gene in families with X-linked hypophosphatemic rickets (HYP)[J]. Hum Mol Genet, 1997, 6 (4): 539–549. DOI:10.1093/hmg/6.4.539 |
| [23] | Raeder H, Shaw N, Netelenbos C, et al. A case of X-linked hypophosphatemic rickets:complications and the therapeutic use of cinicalcet[J]. Eur J Endocrinol, 2008, 159 (Suppl 1): S101–S105. |