 2017, Vol. 19
2017, Vol. 19


朗格罕斯细胞组织细胞增生症(Langerhans cell histiocytosis, LCH)是一种病因不明的来源于骨髓的髓系树状突细胞的克隆增殖性疾病,儿童罕见,年发病率约3~5/百万[1-2]。LCH临床表现差异大,病情轻重不同,国内外治疗方案各异,预后各不相同。初治反应不良、有危险器官受累的多系统LCH患者死亡率高达40%;无危险器官受累的LCH虽致命概率小,但仍有相当比例的恶化、复发(复发率20%~30%)和永久后遗症发生[3]。当今LCH治疗的挑战是降低死亡率,减少复发和永久后遗症的发生。成都市妇女儿童中心2005~2016年间收治LCH儿童患者34例,时间跨度长,经历了诊疗标准的变迁。为了探讨临床表现、分组和治疗方案的决策对预后的影响,进一步提高LCH诊治水平,本文就这部分病例进行回顾性分析,报告如下。
1 资料与方法 1.1 研究对象2005年4月至2016年11月我院儿科确诊LCH初发儿童患者34例,中位年龄14.5个月(22 d至60个月),其中0~2岁23例、 > 2岁11例,男:女为1.83 : 1。
1.2 诊断及疗效判断根据国际组织细胞协会1987年发布的LCH病理诊断标准和2009年的LCH治疗与评估指南[4-5]进行诊断。所有患者均经临床、病理活检和免疫组化染色确诊,并分为单系统LCH(single-system langerhans cell histiocytosis, SS-LCH)12例和多系统LCH(multi-system langerhans cell histiocytosis, MS-LCH)22例。将MS-LCH合并危险器官受累者(肝、脾、肺、血液造血异常)定义为高危组(17例),无危险器官受累的定义为低危组(17)。LCH疗效判断参照1991年国际组织细胞协会制定的LCH疗效判定标准[6]。治愈:症状和/或客观征象完全消失。好转:症状和/或客观征象消退,无新病灶出现。稳定:症状和/或客观征象持续存在,但无新病灶出现。进展或恶化:症状和/或客观征象有进展、新病灶或旧病灶复发。治愈及好转判定为有效。复发指病情治愈、好转或稳定3个月后出现进展或恶化。
1.3 治疗2012年以前确诊的病例共16例,采用LCH-Ⅱ方案治疗[4],6周治疗无效者按原诱导方案再诱导6周。低危组患者总疗程6个月,高危组总疗程12个月;疾病进展或反复者,换用含氨甲喋呤的LCH-Ⅲ方案补救。
14例2012年以后确诊患者的治疗参考LCH 2012年诊疗指南[5]的一线治疗方案,评估稳定或进展者换用二线化疗或DAL-HX90方案。
4例患儿确诊后未行化疗,其中2例为高危患者;1例为新生儿LCH(皮肤受累);1例为锁骨孤立病变,仅行病灶刮除术。
1.4 随访34例患儿中32例接受随访(2例高危患儿失访),随访截至2016年11月,中位随访时间为69(2~130)个月。
累积生存时间即患者确诊之日至死亡或最后随访的时间。无事件生存时间即患儿确诊之日至疾病进展或复发、死亡的时间。
1.5 统计学分析使用SPSS 17.0软件进行数据处理。非正态分布的计量资料用中位数(范围)表示,组间比较采用秩和检验;计数资料用百分率表示,率的比较用Fisher确切概率法。采用Kaplane-Meier分析法进行生存分析,组间生存率的比较采用log-rank检验。P < 0.05为差异有统计学意义。
2 结果 2.1 一般资料高危LCH 17例,男12例、女5例,男:女为2.4 : 1;低危组17例,男10例、女7例,男:女为1.43 : 1,两组的性别构成比差异无统计学意义(P=0.36)。高危组发病中位年龄13(6~20)个月,低危组发病中位年龄36(1~60)个月,以低危组发病年龄较大(Z=-2.986,P < 0.05)。
2.2 临床表现34例患者中皮肤受累者17例,表现为病程迁延、以躯干、颜面部为主的新旧不一的湿疹样皮疹伴色素脱失斑,重者手足心皮疹并伴有坏死。表现为软组织包块者6例,均伴有邻近骨骼的破坏,其中3例以颞部包块起病,1例以枕部包块起病,1例椎旁包块,1例颌部包块。骨受累18例,部分以非外伤后骨折起病,多侵犯颅骨、扁骨、椎骨,四肢长骨受累较少,其中多部位骨破坏者13例、孤立骨受累5例;X片、CT或PET扫描均表现为骨质溶骨性破坏、缺损。垂体受累3例,其中2例表现为尿崩症,1例表现为生长发育迟缓。淋巴结受累6例。耳受累4例,表现为反复溢脓和外耳湿疹样皮疹。肺部受累者9例,表现为咳嗽、伴或不伴气促及肺部喘鸣,影像学表现为肺部广泛磨玻璃样改变或囊泡样病变,合并感染时呈肺实质性病变;其中肺功能受损3例,其2例为阻塞性通气功能障碍、FEV1/FVC降低,1例换气功能障碍、一氧化碳弥散量下降。肝脏受累11例,其中3例发生低蛋白水肿,2例发生黄疸。脾脏肿大者12例(肋下2~6 cm),尚无明显脾功能亢进症状。血液系统受累11例,血象至少2系同时受累,表现为8例贫血(重度贫血3例,中度3例,轻度2例),6例血小板减少,5例中性粒细胞减少;骨髓形态无特异性,多表现为增生活跃或极度活跃,2例可见2%组织细胞;2例并发伊文氏综合征。
2.3 疗效评价34例患者中30例接受化疗,随访时间均超过1年。化疗6周评估总有效率67%(20/30),其中好转20例,稳定2例,进展、恶化8例。化疗12个月评估,总有效率87%(26/30),其中治愈18例、好转或疾病消退8例、进展或恶化4例(2例于化疗期间死亡:1例死于再诱导化疗的第3周,1例死于再诱导化疗后3个月)。
低危组患儿15例,化疗6周评估的有效率为87%(13/15),好转13例,稳定1例,进展、恶化1例;化疗12个月评估的有效率为93%(14/15),其中治愈10例、好转4例、进展或恶化1例。高危组患儿15例,化疗6周有效率为47%(7/15),低于低危组患者(P=0.025),其中好转7例、稳定1例、进展或恶化7例;化疗12个月有效率为80%(12/15),与低危组的差异无统计学意义(P=0.299),其中治愈8例、好转4例、进展或恶化3例。
肺部受累9例,其中危险器官仅肺部受累3例,6周诱导化疗全部有效,至随访末期均无事件存活;另6例至少合并其他1项危险器官受累,6周化疗有效率为67%,至随访末期死亡2例、复发2例。
骨受累LCH 18例,16例接受化疗,其中3例总疗程6个月、13例疗程12个月;6周治疗有效率69%,12个月治疗有效率94%。
2.4 随访34例患者中30例接受化疗并随访;2例高危患儿未行治疗并失访;1例新生儿LCH(皮肤受累)仅予以随访观察,1例锁骨孤立病变者仅行病灶刮除术。随访至截止时间,治愈22例,好转3例,复发4例,死亡4例(1例为复发后死亡),复发、死亡病例均出现在高危组。
4例复发患儿中,1例于总疗程结束1个月后复发,表现为骨等多系统受累伴危险器官损害,家属放弃治疗,3个月后死亡;2例于停药随访期间出现骨质破坏加重,不伴危险器官受损,改用含阿糖胞苷的方案补救治疗后1例好转,无效的1例换用克拉屈滨(2-chlorodeoxyadenosine, 2-CdA)补救治疗后获得完全缓解(目前停药6~12个月无复发);1例病初有4个危险脏器受累的患者停药3个月后肺部病变(双肺弥漫间质改变,叶间隔、小叶间隔不均匀增厚,结节样病变增多、融合,肺功能检测提示通气功能下降)加重,但其余受累器官均评估为好转或消退,未行化疗,目前处于疾病稳定状态、肺部CT无继续加重。
死亡4例,其中复发后死亡1例,2例分别在病程的第3、6个月疾病恶化、死亡;1例先后接受包括长春新碱、甲基泼尼松、依托泊苷、巯基嘌呤、阿糖胞苷和克拉屈滨等多药联合化疗,疾病反复,于病程第20个月死亡。
至随访末期,3例垂体受累的患儿中表现为尿崩症的2例症状无缓解;表现为生长发育迟缓的1例症状缓解,至随访期末身高、体重达同龄儿的P25~P75。
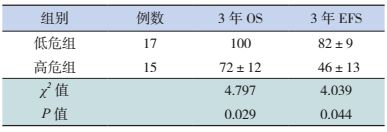
32例患者的3年OS率为86%±6%,3年EFS率为64%±9%。低危组的3年OS率和3年EFS率均高于高危组(P < 0.05),见表 1、图 1。
| 表 1 两组患者预后的比较 (x±s,%) |

|
图 1 高危组、低危组3年总体生存、无事件生存曲线 |
3 讨论
儿童LCH临床表现多样、病变分布广泛,既可以发生单器官、单部位病变,也可发生多器官、多部位病变。本文中LCH患者以皮疹、骨受累、包块为首发症状的比例较高,较早得到诊断,与这些部位的病变容易引起家长和医生的警惕,而且容易获得病理结果有关;而以淋巴结肿大、外耳(中耳)炎、贫血、肝脾受累、尿崩起病的相对较少,诊断时往往有多系统、多器官受累,可能与这些部位受累的症状不典型或病理标本不易获取有关。因此,对这部分症状表现的患者,应注意鉴别LCH,进一步寻找皮肤、骨骼受累的证据、必要时放宽活检指针以获得早期诊断。LCH患儿颅面骨受累可增加尿崩症、垂体前叶激素缺陷和神经系统病变的风险[7-8]。本研究颅面部骨损害不少见,但未见到以促性腺激素分泌异常和以震颤、步态异常、共济失调、构音障碍、头痛、视力障碍、认知和行为异常、精神症状等神经系统退行性损害为首发表现的患者,可能与这些症状的患者未能及时得以与LCH鉴别有关。故对于以尿崩症、生长发育迟缓、性发育迟缓、小脑病变、神经退行性病变等形式起病的患者,要注意追踪肺、血液、骨骼等相关检查及定期监测头颅MRI,以获得LCH诊断的相关证据。
起病年龄、6周诱导治疗的有效性以及危险器官受累是影响LCH预后的重要因子,尤其后两项[2]。本研究高危组患者的发病年龄较低危组更小,6周诱导治疗有效率更低,3年OS和EFS均更低,与之相符。近年来多系统受累LCH患者的多因素分析显示,肺部受累并非独立预后因素,有肺部受累患者的5年总生存率为94%,无肺部受累患者的5年总生存率为96%[9]。本研究将肺部受累LCH归入高危组,提高化疗的强度,单纯肺部受累患儿的6周诱导化疗有效率,以及复发和死亡发生比例均好于合并其他危险器官损害的肺部受累者。提示对于单纯肺部受累的MS-LCH患儿,降低化疗强度也有可能达到良好预后。
低危LCH的骨病变最常见。对危及中枢神经系统的单灶骨病变、单系统多灶骨病变或多系统骨病变患者,国际上常规采用长春花碱和强的松联合化疗,并强调疗程要足够长,推荐12个月疗程,可以明显降低复发率,并降低患尿崩症和其他后遗症的风险[10-11]。而对复发、难治的骨病变或难治的低危、多系统LCH补救治疗推荐克拉屈滨和帕米膦酸二钠等双膦酸盐[12-13]。本案骨受累患儿12个月治疗有效率达93.7%,即使复发,只要不合并危险器官受累,再化疗仍然获得良好预后。另外我们注意到,骨骼受累在影像学上恢复较慢,6周评估时多数缺损部位未见到明显缩小,但其他器官系统病变好转,进入维持治疗后半年至1年才出现X片或CT明显好转。因此对于骨损害在6~12周化疗后无明显变化者,不推荐增加化疗强度或更换治疗策略。
对高危组患者,国际组织细胞疾病协会进行的前瞻性研究LCH-Ⅰ、LCH-Ⅱ、LCH-Ⅲ表明,增加治疗强度、延长初始治疗时间和维持治疗时间,可以显著提高化疗有效率,降低死亡率和复发率[14]。国内也有采用改良LCH-Ⅱ、改良LCH-Ⅲ、改良DAL-HX83/90等方案治疗的单中心报道[15-17]。而对难治性高危组患者,可采用如2-CDA、2-CDA联合阿糖胞苷等方案挽救治疗[13, 18]。本案高危组患者即使增加了诱导治疗的强度,6周诱导治疗有效率仍然不足50%,但治疗12个月评估,疗效提高了近1倍。可能6周进行评估相对较早。另外,对于延长初始治疗疗程无好转或诱导治疗6周后病情进展的,更换治疗方案可使预后得到改善。有研究报道,BRAFV600E突变与儿童高危LCH有密切关系,突变阳性者更多地表现出早期化疗反应差和永久性损害发生率高,提示应用BRAF抑制剂或联合应用BRAF抑制剂和MEK抑制剂(已使用于成人)可能是儿童高危LCH的有效补救治疗[19-21]。本案中4例死亡患儿均有危险器官受累,年龄均小于18个月,均接受标准一线化疗和2种以上的补救治疗方案无效,若能接受BRAFV600E突变检测,也许可通过靶向治疗获得挽救。
近年来儿童LCH的疗效和预后得到了很大程度改善[15-17, 22],诱导治疗有效率提高到70%~80%,5年存活率达到80%以上,复发率降至25%~30%,死亡率降至10%~20%。本研究诱导化疗总有效率接近70%,1年缓解率(有效率)达90%以上,3年存活率达82%左右。疗效基本达到上述水平。但高危组儿童仍有较高的难治和复发、死亡比例,低危组如出现尿崩症,也难以通过化疗改善;另一方面,对于单纯肺部受累和骨损害影像学恢复较慢的LCH患儿,降低化疗强度或延长观察时间可避免过度治疗。因此,需要进一步开展多中心临床研究,以寻求LCH个体化的治疗方案及靶向治疗措施改善预后。
| [1] | Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells[J]. J Immunol, 2010, 184 (8): 4557–4667. DOI:10.4049/jimmunol.0902336 |
| [2] | Minkov M. Multisystem Langerhans cell histiocytosis in children:current treatment and future directions[J]. Paediatr Drugs, 2011, 13 (2): 75–86. DOI:10.2165/11538540-000000000-00000 |
| [3] | Haupt R, Nanduri V, Calevo MG, et al. Permanent consequences in Langerhans cell histiocytosis patients:a pilot study from the Histiocyte Society-Late Effects Study Group[J]. Pediatr Blood Cancer, 2004, 42 (5): 438–444. DOI:10.1002/(ISSN)1545-5017 |
| [4] | 高举, 袁粒星. 朗格汉斯细胞组织细胞增生症的诊断和治疗[J]. 实用儿科临床杂志, 2006, 21 (15): 1037–1040. DOI:10.3969/j.issn.1003-515X.2006.15.045 |
| [5] | 吴升华. 朗格汉斯细胞组织细胞增生症评估与治疗指南介绍[J]. 中华儿科杂志, 2012, 50 (2): 155–158. |
| [6] | Gadner H, Grois N, Arico M, et al. A randomized trial of treatment for multisystem Langerhans' cell histiocytosis[J]. J Pediatr, 2001, 138 (5): 728–734. DOI:10.1067/mpd.2001.111331 |
| [7] | Grois N, Pötschger U, Prosch H, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis[J]. Pediatr Blood Cancer, 2006, 46 (2): 228–233. DOI:10.1002/(ISSN)1545-5017 |
| [8] | Fahrner B, Prosch H, Minkov M, et al. Long-term outcome of hypothalamic pituitary tumors in Langerhans cell histiocytosis[J]. Pediatr Blood Cancer, 2012, 58 (4): 606–610. DOI:10.1002/pbc.v58.4 |
| [9] | Ronceray L, Pötschger U, Janka G, et al. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis:effect on course and outcome[J]. J Pediatr, 2012, 161 (1): 129–133. DOI:10.1016/j.jpeds.2011.12.035 |
| [10] | Gadner H, Heitger A, Grois N, et al. Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX-83 Study Group[J]. Med Pediatr Oncol, 1994, 23 (2): 72–80. DOI:10.1002/(ISSN)1096-911X |
| [11] | Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis[J]. Blood, 2013, 121 (25): 5006–5014. DOI:10.1182/blood-2012-09-455774 |
| [12] | Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate[J]. J Pediatr Hematol Oncol, 2001, 23 (1): 54–56. DOI:10.1097/00043426-200101000-00013 |
| [13] | Weitzman S, Braier J, Donadieu J, et al. 2'-Chlorodeoxyadeno-sine (2-CdA) as salvage therapy for Langerhans cell histiocytosis (LCH) results of the LCH-S-98 protocol of the Histiocyte Society[J]. Pediatr Blood Cancer, 2009, 53 (7): 1271–1276. DOI:10.1002/pbc.v53:7 |
| [14] | Gadner H, Grois N, Pötschger U, et al. Improved outcome in multisystem Langerhans cell histiocytosis is associated with therapy intensification[J]. Blood, 2008, 111 (5): 2556–2562. DOI:10.1182/blood-2007-08-106211 |
| [15] | 吴方方, 高怡瑾, 潘慈, 等. 儿童多脏器受累朗格汉斯细胞组织细胞增生症131例临床研究[J]. 中华儿科杂志, 2016, 54 (5): 349–353. |
| [16] | 宋爱琴, 李学荣, 庞秀英, 等. 儿童朗格汉斯细胞组织细胞增生症26例临床分析[J]. 临床儿科杂志, 2011, 29 (5): 431–434. |
| [17] | 黄俊彬, 江莉, 陈纯, 等. 改良DAL-HX83/90和JLSG-96方案治疗儿童朗格汉斯细胞组织细胞增生症的疗效比较[J]. 中国实验血液学杂志, 2016, 24 (4): 1190–1195. |
| [18] | Bernard F, Thomas C, Bearand Y, et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction[J]. Eur J Cancer, 2005, 41 (17): 2682–2689. DOI:10.1016/j.ejca.2005.02.007 |
| [19] | Héritier S, Emile JF, Barkaoui MA, et al. BRAF mutation correlates with high-risk Langerhans cell histiocytosis and increased resistance to first-line therapy[J]. J Clin Oncol, 2016, 34 (25): 3023–3030. DOI:10.1200/JCO.2015.65.9508 |
| [20] | Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma[J]. N Engl J Med, 2014, 371 (20): 1867–1876. DOI:10.1056/NEJMoa1408868 |
| [21] | Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma:a multicentre. double-blind, phase 3 randomised controlled trial[J]. Lancet, 2015, 386 (9992): 444–451. DOI:10.1016/S0140-6736(15)60898-4 |
| [22] | 泥永安, 孙立荣. 朗格汉斯细胞组织细胞增生症治疗进展[J]. 临床儿科杂志, 2015, 33 (3): 291–293. |