 2017, Vol. 19
2017, Vol. 19


 , LI Lan, YU Xiao-Jun, KE Jiang-Wei, HE Mei-Juan, ZHOU Hong-Ping, YANG Wen-Ping, WANG Wen-Xing
, LI Lan, YU Xiao-Jun, KE Jiang-Wei, HE Mei-Juan, ZHOU Hong-Ping, YANG Wen-Ping, WANG Wen-Xing
金黄色葡萄球菌(Staphylococcus aureus, SA)是一种具有多种毒力因子的革兰阳性球菌[1],是儿童社区及医院感染的重要病原菌,近年来感染率逐年增高[2]。其中尤其是耐甲氧西林SA感染严重危害了儿童的身体健康。
SA能够通过直接对抗吞噬作用[3]、抑制补体介导的调理作用[4-5]、免疫杀伤作用[6]和竞争性抑制趋化作用[7]来对抗人体的免疫防御。还能通过腺苷合成酶(ADSA)接受机体感染葡萄球菌过程中的分子信号,使细胞外腺苷总量急剧增加,降低抗菌肽对它的识别[8]。并且,SA能够分泌具有强抗氧化性的类胡萝卜素,加强其抗氧化能力[9]。SA在逃避宿主免疫反应的同时,还能不断释放毒素及侵袭性酶类,是造成慢性迁徙性感染的主要原因[10]。
重组融合蛋白IL-18由小鼠IL-18重组表达质粒并应用原核表达系统获得,具有生物活性。有研究指出,IL-18能够上调干扰素-γ(interferon-γ, IFN-γ)介导的细胞免疫反应,增加血清中肿瘤坏死因子(tumor necrosis factor, TNF)、粒细胞集落刺激因子(granulocyte colony stimulating factor, G-CSF)、巨噬细胞炎性蛋白(macrophage inflammatory protein, MIP)的水平,以及增加/恢复中性粒细胞计数,从而提高中性粒细胞功能,增强细胞的吞噬活性,对胞外菌进行吞噬[11];而且能够上调IgM抗体介导的体液免疫应答和中性粒细胞介导的免疫反应,增加免疫细胞的募集和活化,增强补体介导的调理作用,进一步对胞外菌进行破坏,并协同细胞免疫反应对胞外菌进行进一步吞噬[12]。
目前江西省地区儿童SA感染的耐药率逐年上升,本课题组前期对2009年1月至2012年12月在江西省儿童医院住院的SA侵袭性感染患儿分离的161株SA进行了研究,发现SA对青霉素及红霉素有较高的耐药性,耐甲氧西林SA的检出率逐年升高[13]。重组融合蛋白IL-18作为一种有活性的细胞因子,能否成功通过鼻腔免疫进入机体,干预SA对免疫的防御机制,以降低SA的致病力、耐药率和儿童的病死率?本研究旨在通过观察重组融合蛋白IL-18干预后SA所致的免疫相关炎性因子的变化,了解重组融合蛋白IL-18在体内防御SA感染的可能机制。
1 材料与方法 1.1 实验动物6~8周龄SPF级雌性BLAB/c小鼠40只,体重16~18 g,购于江西中医学院实验动物中心。在室温下分笼饲养,每只鼠笼安置5只小鼠,进食平衡饲料,自由饮水,光线充足,空气中相对湿度约为47%,室内保持通风。
1.2 主要试剂重组融合蛋白IL-18(北京义翘神州生物技术有限公司)、IL-4、IFN-γ、TNF、G-CSF、IgM ELISA试剂盒(武汉Elabscience生物科技有限公司)、总RNA提取试剂盒、逆转录及RT-qPCR试剂盒(美国Promega公司)、PCR引物(上海捷瑞生物公司)、SA标准株(ATCC-29213)(江西乐悠生物科技有限公司)。
1.3 实验动物分组将40只小鼠随机分为对照组、SA感染组、IL-18免疫组(简称免疫组)、SA感染+IL-18干预组(简称干预组);每组10只。
1.4 实验动物模型的建立参照文献[14]建立动物模型。对照组:实验第1周每天每只小鼠20 μL PBS滴鼻,于实验第8天继续以20 μL PBS滴鼻。SA感染组:实验第1周每天每只小鼠20 μL PBS滴鼻,于实验第8天鼻腔接种1.5×106 CFU/mL SA液20 μL。免疫组:实验第1周每天每只小鼠20 μL重组融合蛋白IL-18滴鼻,于实验第8天以20 μL PBS滴鼻。干预组:实验第1周每天每只小鼠20 μL重组融合蛋白IL-18滴鼻,于实验第8天鼻腔接种1.5×106 CFU/mL SA液20 μL。
1.5 小鼠全血标本的采集及方法感染后第3天采用戊巴比妥钠腹腔注射将小鼠麻醉,仰卧固定于实验台上,于颈部正中作一长约3 cm的纵行切口,钝性分离左侧颈总动脉,穿刺取各组小鼠动脉血3 mL,置于普通管中,放置于4℃冰箱中备用。
1.6 小鼠支气管肺泡灌洗液标本的获取采集小鼠血液标本后,以Y型管作为气管插管,以400 μL生理盐水灌洗全肺,每只小鼠回收支气管肺泡灌洗液(BALF)约1.2 mL。
1.7 小鼠肺组织标本的获取收集完小鼠血液及BALF标本后,此时小鼠仍处于麻醉状态,迅速打开胸腔,分离出小鼠肺组织,用无菌器械剪切成任何一边均未超过0.5 cm的组织块,放入预先盛有RNAlater样本保存液的标本留取瓶中,置于4℃冰箱中过夜,让样本保存液充分渗透入组织块中,后放入-20℃冰箱中长期冻存,用于RT-qPCR检测肺组织中MIP-1α和MIP-2β mRNA的含量。
1.8 ELISA法测定小鼠血清及BALF中IL-4、IFN-γ、TNF、G-CSF、IgM含量取3 mL全血经3 000 r/min离心10 min后分离出血清,置于-80℃冰箱中备用。实验开始前将各组血清及各试剂平衡至室温,提前打开酶标仪电源,设置好检测程序,预热仪器,450 mm波长测量各孔的光密度值(OD值)。按照试剂盒使用说明书配置所需试剂,严格按照操作步骤分别测定小鼠血清中IL-4、IFN-γ、TNF、G-CSF、IgM的含量。
1.9 RT-qPCR法测定小鼠肺组织中MIP-1α和MIP-2β mRNA水平取171 mg肺组织块加入1 mL RNA组织裂解液,按照组织总RNA提取试剂盒说明书进行总RNA提取。取总RNA 2 μL,按照逆转录试剂盒说明书合成cDNA后进行荧光定量PCR扩增。反应体系(50 μL):cDNA 10 μL,GoTaq qPCR Master Mix 25 μL,上下游引物各1 μL,Nucleasa-free水13 μL。反应条件:95℃ 1 min;94℃ 15 s,57℃ 30 s,72℃ 45 s,40个循环;再接融解曲线。引物序列及扩增片段长度见表 1。
| 表 1 荧光定量PCR检测用引物序列 |
1.10 统计学分析
采用SPSS 19.0统计软件对实验数据进行统计学分析。符合正态分布的计量资料用均数±标准差(x±s)表示,多组间比较采用单因素方差分析,组间两两比较采用SNK-q检验,P < 0.05为差异有统计学意义。
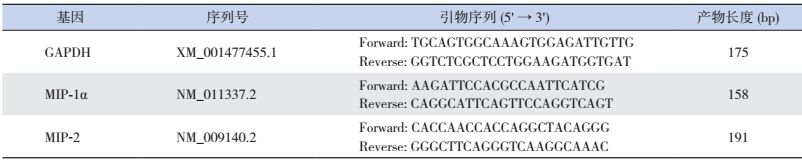
2 结果 2.1 重组融合蛋白IL-18对小鼠血清及BALF中IL-4水平的影响SA感染组、免疫组及干预组小鼠血清及BALF中IL-4水平均较对照组增高(P < 0.05)。干预组小鼠血清及BALF中IL-4水平较SA感染组增高(P < 0.05)。SA感染组小鼠血清及BALF中IL-4水平较免疫组增高(P < 0.05)。见表 2。
| 表 2 各组小鼠血清及BALF中IL-4水平比较 (x±s,pg/mL) |
2.2 重组融合蛋白IL-18对小鼠血清及BALF中IFN-γ水平的影响
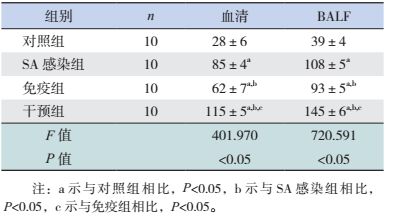
SA感染组、干预组小鼠血清IFN-γ水平均较对照组降低(P < 0.05);免疫组小鼠血清IFN-γ水平较对照组、SA感染组、干预组均增高(P < 0.05);干预组小鼠血清IFN-γ水平较SA感染组增高(P < 0.05)。免疫组、干预组小鼠BALF中IFN-γ水平较对照组和SA感染组均增高(P < 0.05);SA感染组小鼠BALF中IFN-γ水平较对照组降低(P < 0.05);免疫组小鼠BALF中IFN-γ水平低于干预组(P < 0.05)。见表 3。
| 表 3 各组小鼠血清及BALF中IFN-γ水平比较 (x±s,pg/mL) |
2.3 重组融合蛋白IL-18对小鼠血清及BALF中TNF水平的影响
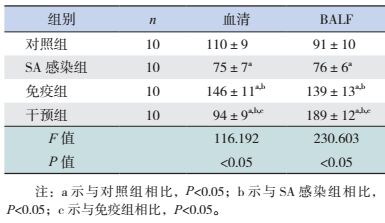
SA感染组、干预组小鼠血清及BALF中TNF水平均较对照组和免疫组增高(P < 0.05)。免疫组小鼠血清及BALF中TNF水平与对照组比较差异无统计学意义(P > 0.05)。干预组小鼠血清及BALF中TNF水平较SA感染组均降低(P < 0.05)。见表 4。
| 表 4 各组小鼠血清及BALF中TNF水平比较 (x±s,pg/mL) |
2.4 重组融合蛋白IL-18对小鼠血清及BALF中G-CSF水平的影响
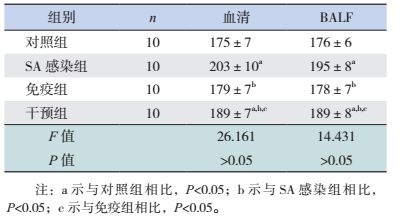
SA感染组、免疫组及干预组小鼠血清及BALF中G-CSF浓度均较对照组明显增高(P < 0.05)。干预组小鼠血清及BALF中G-CSF浓度较SA感染组和免疫组增高(P < 0.05)。SA感染组小鼠血清及BALF中G-CSF浓度较免疫组增高(P < 0.05)。见表 5。
| 表 5 各组小鼠血清及BALF中G-CSF水平比较 (x±s,pg/mL) |
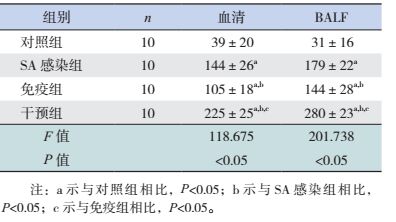
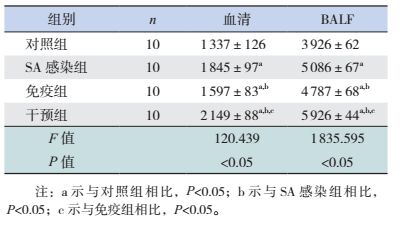
2.5 重组融合蛋白IL-18对小鼠血清及BALF中IgM水平的影响
SA感染组、免疫组及干预组小鼠血清及BALF中IgM浓度均较对照组升高(P < 0.05)。干预组小鼠血清及BALF中IgM浓度均较SA感染组和免疫组升高(P < 0.05)。SA感染组小鼠血清及BALF中IgM浓度较免疫组升高(P < 0.05)。见表 6。
| 表 6 各组小鼠血清及BALF中IgM水平比较 (x±s,pg/mL) |
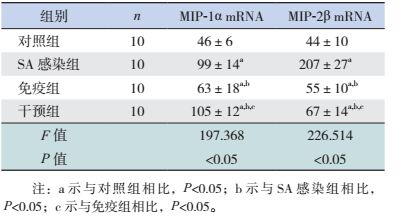
2.6 重组融合蛋白IL-18对小鼠肺组织中MIP-1α和MIP-2β mRNA水平的影响
SA感染组、干预组及免疫组小鼠肺组织中MIP-1α及MIP-2β含量均较对照组增高(P < 0.05)。免疫组小鼠肺组织中MIP-1α及MIP-2β含量均较SA感染组和干预组降低(P < 0.05)。SA感染组MIP-1α含量低于干预组,而MIP-2β含量高于干预组(P < 0.05)。见表 7。
| 表 7 各组小鼠肺组织中MIP-1α和MIP-2β mRNA水平比较 (x±s) |
3 讨论
SA作为一种共生菌,能够造成人体不同部位同时感染。鼻前庭是SA最常见的菌群定值部位,约20%人口鼻前庭菌群定值是永久的[15-16]。通常定植于鼻前庭的SA并不致病,但当机体免疫功能下降时,SA将透过黏膜防御体系,引起包括各类皮肤软组织感染和致死性的感染性疾病。因而机体的免疫系统对病原体的识别和清除功能对预防SA感染具有重要意义。
Th1和Th2细胞可释放IFN-γ和IL-4两种炎症介质,两者表达水平的变化可间接反映机体的免疫状态及机体感染后炎症的发展趋势,而两者的分泌失衡也能导致气道炎性损伤。IL-18作为一种促炎性细胞因子,本身能诱导表达IL-18受体(IL-18R)的细胞产生CXC和CC型化学趋化因子,引起巨噬细胞、单核细胞、成纤维细胞释放IL-18[17]。而IL-18的释放能进一步诱导Th1细胞和NK细胞产生IFN-γ[18]。IFN-γ能在炎症反应中增加天然免疫细胞的杀伤活性[19],但当IFN-γ分泌不足时能进一步加重气道的炎性损伤[20]。本研究中,SA感染组、干预组小鼠血清IFN-γ虽较对照组降低,但免疫组小鼠血清IFN-γ较对照组增高。而在BALF中,免疫组、干预组小鼠BALF中IFN-γ均较对照组增高,且免疫组小鼠BALF中IFN-γ较SA感染组增高。由此可见重组融合蛋白IL-18能够增加IFN-γ在血清及BALF中的浓度,维持IFN-γ与其他细胞因子的平衡,进一步增加机体感染SA后免疫细胞的杀伤活性,这与国外的相关研究一致[21]。IL-4能够调控巨噬细胞释放细胞因子,增加体内巨噬细胞吞噬SA的能力[19]。SA感染组、免疫组、干预组小鼠血清及BALF中IL-4浓度均较对照组增高。且SA感染组小鼠血清及BALF中IL-4浓度较免疫组增高。说明重组融合蛋白IL-18不仅能提高IL-4在血清及BALF中的浓度,并且能够降低SA感染后小鼠血清及BALF中的IL-4浓度,使其与IFN-γ维持稳态,协同增加机体对SA的杀伤作用。
IL-18能够通过MyD88信号参与IgM、IgG等免疫球蛋白的产生环节,并且能够激活免疫系统,早期产生抗体反应,进一步的释放IgM、IgG[22]。本研究中,免疫组和干预组小鼠血清及BALF中IgM浓度均较对照组增高。TNF参与机体抗菌防御的多种机制,包括对中性粒细胞招募,增强中性粒细胞对血管的附着力[23];有研究表明,在机体感染SA早期,TNF能够特异性的识别SA抗原,激活机体的免疫系统引起TNF释放TNFR1受体,从而进一步增加体内TNF含量,增强机体对抗炎性反应的初始阶段[24]。当TNF水平达到一个峰值,能诱发气道上皮细胞产生大量细胞因子[25],TNF在清除细菌的过程中起着至关重要的作用,但过高水平的TNF可能导致组织损伤、器官衰竭和死亡[26]。抑制内源性TNF能增加SA感染的病死率[27]。本研究中,SA感染组和干预组小鼠血清及BALF中TNF浓度均较对照组增高,但免疫组血清及BALF中TNF浓度均较对照组无明显改变,干预组小鼠血清及BALF中TNF均较SA感染组降低。这表明IL-18能够增高SA感染后机体血液及BALF中TNF的浓度,从而加强对SA的清除作用,也与国外的相关报道一致[28]。
MIP-1α、MIP-2β分属于趋化因子CC家族和CXC家族,IL-18能够诱导IL-18R表达MIP-1α、MIP-2β,从而明显加强炎症免疫反应细胞的趋化活性,进一步调节效应细胞的功能。MIP-1α能作用于NK细胞,从而增加肺内巨噬细胞对SA的吞噬功能;MIP-2β能特异性作用于中性粒细胞,进一步趋化和激活中性粒细胞[29]。G-CSF由巨噬细胞产生,能通过影响细胞表面黏附分子的表达,特异性活化中性粒细胞,进一步趋化炎性反应[30]。本研究中SA感染组、免疫组、干预组小鼠血清及BALF中G-CSF均较对照组明显增高。SA感染组、免疫组、干预组小鼠肺组织中MIP-1α、MIP-2β含量均较对照组增高,这表明重组融合蛋白IL-18能增高SA感染后体内G-CSF的含量以及肺组织内MIP-1α、MIP-2β的mRNA表达水平,从而进一步增加巨噬细胞对SA的吞噬功能,这些结果也与国外的报道一致[31-32]。
本研究发现重组融合蛋白IL-18经鼻腔黏膜免疫可以明显提高TNF、G-CSF、IgM相关炎症因子的表达水平,并且增加MIP-1α、MIP-2β在肺组织中的表达,促进机体抗感染免疫反应,从而增强了机体清除病原体的能力。因而推测,调节IFN-γ、IL-4、G-CSF和IgM等炎症因子表达可能是重组融合蛋白IL-18增强小鼠抗感染能力的重要机制。
| [1] | Chambers HF, Deleo FR. Waves of resistance:Staphylococcus aureus in the antibiotic era[J]. Nat Rev Microbiol, 2009, 7 (9): 629–641. DOI:10.1038/nrmicro2200 |
| [2] | Seil JT, Webster TJ. Antimicrobial applications of nanotechnology:methods and literature[J]. Int J Nanomedicine, 2012, 7 : 2767–2781. |
| [3] | Peterson PK, Verhoef J, Sabath LD, et al. Effect of protein A on staphylococcal opsonization[J]. Infect Immun, 1977, 15 (3): 760–764. |
| [4] | Cunnion KM, Zhang HM, Frank MM. Availability of complement bound to Staphylococcus aureus to interact with membrane complement receptors influences efficiency of phagocytosis[J]. Infect Immun, 2003, 71 (2): 656–662. DOI:10.1128/IAI.71.2.656-662.2003 |
| [5] | Cunnion KM, Lee JC, Frank MM. Capsule production and growth phase influence binding of complement to Staphylococcus aureus[J]. Infect Immun, 2001, 69 (11): 6796–6803. DOI:10.1128/IAI.69.11.6796-6803.2001 |
| [6] | Kitur K, Parker D, Nieto P, et al. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage[J]. PLoS Pathog, 2015, 11 (4): e1004820. DOI:10.1371/journal.ppat.1004820 |
| [7] | de Haas CJ, Veldkamp KE, Peschel A, et al. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent[J]. J Exp Med, 2004, 199 (5): 687–695. DOI:10.1084/jem.20031636 |
| [8] | Thammavongsa V, Kern JW, Missiakas DM, et al. Staphylococcus aureus synthesizes adenosine to escape host immune responses[J]. J Exp Med, 2009, 206 (11): 2417–2427. DOI:10.1084/jem.20090097 |
| [9] | Liu CI, Liu GY, Song Y, et al. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence[J]. Science, 2008, 319 (5868): 1391–1394. DOI:10.1126/science.1153018 |
| [10] | Tuchscherr L, Medina E, Hussain M, et al. Staphylococcus aureus phenotype switching:an effective bacterial strategy to escape host immune response and establish a chronic infection[J]. EMBO Mol Med, 2011, 3 (3): 129–141. DOI:10.1002/emmm.201000115 |
| [11] | Kinoshita M, Shinomiya N, Ono S, et al. Restoration of natural IgM production from liver B cells by exogenous IL-18 improves the survival of burn-injured mice infected with Pseudomonas aeruginosa[J]. J Immunol, 2006, 177 (7): 4627–4635. DOI:10.4049/jimmunol.177.7.4627 |
| [12] | Kuranaga N, Kinoshita M, Kawabata T, et al. Interleukin-18 protects splenectomized mice from lethal Streptococcus pneumoniae sepsis independent of interferon-gamma by inducing IgM production[J]. J Infect Dis, 2006, 194 (7): 993–1002. DOI:10.1086/jid.2006.194.issue-7 |
| [13] | Qiao Y, Ning X, Chen Q, et al. Clinical and molecular characteristics of invasive community-acquired Staphylococcus aureus infections in Chinese children[J]. BMC Infect Dis, 2014, 14 : 582. DOI:10.1186/s12879-014-0582-4 |
| [14] | 陈凌, 郭盛, 郝春莉, 等. 重组融合蛋白白细胞介素-17F/His对肺炎链球菌感染小鼠免疫相关炎性因子表达的影响[J]. 中华实用儿科临床杂志, 2014, 29 (4): 255–260. |
| [15] | Wertheim HF, Melles DC, Vos MC, et al. The role of nasal carriage in Staphylococcus aureus infections[J]. Lancet Infect Dis, 2005, 5 (12): 751–762. DOI:10.1016/S1473-3099(05)70295-4 |
| [16] | Bogaert D, van Belkum A, Sluijter M, et al. Colonisation by Streptococcus pneumoniae and Staphylococcus aureus in healthy children[J]. Lancet, 2004, 363 (9424): 1871–1872. DOI:10.1016/S0140-6736(04)16357-5 |
| [17] | Morel JC, Park CC, Kumar P, et al. Interleukin-18 induces rheumatoid arthritis synovial fibroblast CXC chemokine production through NFkappaB activation[J]. Lab Invest, 2001, 81 (10): 1371–1383. DOI:10.1038/labinvest.3780351 |
| [18] | Yoshimoto T, Tsutsui H, Tominaga K. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine released by basophils[J]. Proc Natl Acad Sci U S A, 1999, 96 (24): 13962–13966. DOI:10.1073/pnas.96.24.13962 |
| [19] | Kang DH, Weaver MT. Airway cytokine responses to acute and repeated stress in a murine model of allergic asthma[J]. Bio Psychol, 2010, 84 (1): 66–73. DOI:10.1016/j.biopsycho.2009.10.005 |
| [20] | Truyen E, Coteur L, Dilissen E, et al. Evaluation of airway inflammation by quantitative Th1/Th2 cytokine mRNA measurement in sputum of asthma patients[J]. Thorax, 2006, 61 (3): 202–208. DOI:10.1136/thx.2005.052399 |
| [21] | Pagès F, Lazar V, Berger A, et al. Analysis of interleukin-18, interleukin-1 converting enzyme (ICE) and interleukin-18-related cytokines in Crohn's disease lesions[J]. Eur Cytokine Netw, 2001, 12 (1): 97–104. |
| [22] | Enoksson, S L, Grasset EK, Hägglöf T, et al. The inflammatory cytokine IL-18 induces self-reactive innate antibody responses regulated by natural killer T cells[J]. Proc Natl Acad Sci U S A, 2011, 108 (51): E1399–E1407. DOI:10.1073/pnas.1107830108 |
| [23] | Lorente JA, Marshall JC. Neutralization of tumor necrosis factor in preclinical models of sepsis[J]. Shock, 2005, 24 (Suppl 1): 107–119. |
| [24] | Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system[J]. Clin Microbiol Rev, 2005, 18 (3): 521–540. DOI:10.1128/CMR.18.3.521-540.2005 |
| [25] | Gómez MI, Lee A, Reddy B, et al. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1[J]. Nat Med, 2004, 10 (8): 842–848. DOI:10.1038/nm1079 |
| [26] | Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis[J]. N Engl Med, 2003, 348 (2): 138–150. DOI:10.1056/NEJMra021333 |
| [27] | Fei Y, Wang W, Kwiecinski J, et al. The combination of a tumor necrosis factor inhibitor and antibiotic alleviates staphylococcal arthritis and sepsis in mice[J]. J Infect Dis, 2011, 204 (3): 348–357. DOI:10.1093/infdis/jir266 |
| [28] | Biet F, Locht C, Kremer L. Immunoregulatory functions of interleukin 18 and its role in defense against bacterial pathogens[J]. J Mol Med (Berl), 2002, 80 (3): 147–162. DOI:10.1007/s00109-001-0307-1 |
| [29] | Lomas-Neira JL, Chung CS, Wesche DE, et al. In vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury[J]. J Leukoc Biol, 2005, 77 (6): 846–853. DOI:10.1189/jlb.1004617 |
| [30] | Semerad CL, Christopher MJ, Liu F, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow[J]. Blood, 2005, 106 (9): 3020–3027. DOI:10.1182/blood-2004-01-0272 |
| [31] | Dallaire F, Ouellet N, Bergeron Y, et al. Microbiological and inflammatory factors associated with the development of pneumococcal pneumonia[J]. J Infect Dis, 2001, 184 (3): 292–300. DOI:10.1086/jid.2001.184.issue-3 |
| [32] | Rijneveld AW, van den Dobbelsteen GP, Florquin S, et al. Roles of interleukin-6 and macrophage inflammatory protein-2 in pneumolysin-induced lung inflammation in mice[J]. J Infect Dis, 2002, 185 (1): 123–126. DOI:10.1086/jid.2002.185.issue-1 |