 2017, Vol. 19
2017, Vol. 19



先天性胆汁酸合成障碍2型(congenital bile acid synthesis defect type 2, CBAS2),又称为原发性Δ4-3-氧固醇5β-还原酶缺陷病,是一种由AKR1D1基因突变引起的常染色体隐性遗传病[1]。其主要的临床表现为胆汁淤积性黄疸,脂肪、脂溶性维生素吸收障碍,血清转氨酶升高,但血清总胆汁酸和γ-谷氨酰转肽酶正常[1]。AKR1D1基因位于染色体7q32-33,全长42 kb,包含9个外显子,编码2.7 kb的mRNA分子,其蛋白质产物为含326个氨基酸的Δ4-3-氧固醇5β-还原酶[2-3]。该酶主要表达在肝细胞,作为一种NADPH依赖性酶,催化所有含3-氧-4烯结构的类固醇激素和胆汁酸前体的还原反应[2-4]。
AKR1D1基因分析是CBAS2确诊的可靠依据。迄今为止,国外文献已报道12种AKR1D1基因突变,包括2种移码突变,2种无义突变和8种错义突变[5-10]。2010年,Zhao等[10]首次在2位中国大陆患者中检测出AKR1D1基因突变,说明我国也存在CBAS2患者;但目前国内涉及本病的文献仅有3篇综述[11-13],CBAS2的临床诊断和治疗经验尚需要进一步积累。本文报道1例CBAS2患儿的临床和AKR1D1基因突变特征,希望作为引玉之砖,为本病确诊提供分子依据,并为后续诊治研究提供参考。
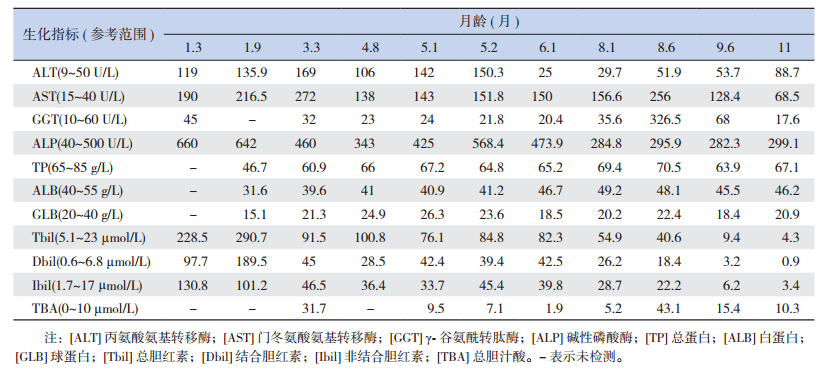
1 资料与方法 1.1 病例介绍患儿,男,8个月4 d,因“全身皮肤、巩膜黄染7月余”入院。患儿生后约半个月开始出现皮肤、巩膜黄染,伴尿黄、陶土样大便,家属未予重视。生后1个月8 d时到当地医院就诊,查肝功能:总胆红素228.5 μmol/L,结合胆红素97.7 μmol/L,非结合胆红素130.8 μmol/L,丙氨酸氨基转移酶119 U/L,门冬氨酸氨基转移酶190 U/L,γ-谷氨酰转肽酶45 U/L。后复查肝功能仍异常,但病因不明(表 1)。3个月10 d时在另一医院行“腹腔镜探查+胆道探查+胆道造影+胆道冲洗+肝活检术”,肝脏病理活检示:肝细胞明显增大,可见大量多核巨肝细胞和明显淤胆,肝窦稍扩张充血,门管区小胆管和纤维组织轻度增生,呈胆汁淤积性肝炎病理改变(图 1)。诊断为“胆汁淤积性肝病”,并给予百佳力(无乳糖中链甘油三酯配方奶粉)喂养及口服“联苯双酯”等药物,黄疸逐渐减轻,但肝功能异常持续(表 1)。为进一步诊治而至我院儿科就诊。患儿自起病以来,精神、睡眠尚可,胃纳差,米糊或奶粉每日约600 mL,大便为黄色,小便正常,体重增长缓慢。
| 表 1 患儿历次生化检查结果 |

|
图 1 患儿肝脏组织学检查结果 (A~B:苏木精-伊红染色,C:免疫组化,D:Masson染色,×200)可见肝小叶结构絮乱,肝细胞胞质呈细颗粒样或“毛玻璃”状,可见多核巨细胞,肝细胞淤胆(A),同时细胞间质(A)和汇管区(B)有大量淋巴细胞浸润;CK8蛋白免疫组化染色显示汇管区边缘小胆管轻度增生(C);而Masson染色显示汇管区和细胞间质纤维组织增生(D)。以上改变,符合胆汁淤积性肝炎病理特征。 |
患儿系第2胎第2产,出生胎龄40周,经阴道分娩出生,出生体重2.85 kg,身长47.0 cm,出生时无窒息史。父母体健,非近亲结婚。否认家族类似病史及传染病史。
体格检查:体重6.85 kg( < -2 SD),身长65 cm( < -3 SD),头围42 cm( < -2 SD)。神志清楚,精神可,发育、营养差。全身皮肤、巩膜轻度黄染,无皮疹及皮下出血点,浅表淋巴结未触及肿大。前囟平软,大小约1 cm×1 cm。唇红,咽无充血,未见脓性分泌物。颈软,无抵抗,气管居中,胸廓对称,无明显畸形,三凹征(-),双肺呼吸音清,未闻及干、湿性罗音。心律齐,心音有力,未闻及病理性杂音。腹稍饱满,肝右肋下8 cm,质地中等,脾肋下未及,移动性浊音(-),肠鸣音正常。脊柱、四肢无畸形,肛门及外生殖器无异常。腹壁、膝腱和跟腱等生理反射可引出,克氏、布氏和巴氏征均阴性。
辅助检查:入院后查血常规大致正常。生化检查发现肝功能异常(表 1)。血清锌4.9 µmol/L(参考值11.47~25.5 µmol/L),25-(OH)D 5.89 ng/mL(参考值30~100 ng/mL)。
1.2 代谢性肝病组套二代测序用EDTA抗凝管收集患儿静脉血2 mL,并用Blood DNA Mini kit试剂盒(杭州新景生物试剂开发有限公司)提取患儿基因组DNA 5 μg。Covaris S2超声仪将基因组DNA打断至约150 bp片段长度,随后片段化DNA经末端补平修复,两端加A,连接adaptor,扩增和纯化后,与探针混于杂交体系中,建立含有与代谢性肝病相关基因(JAG1、NOTCH2、ATP8B1、ABCB11等249个基因)的全基因组文库。用液相捕获试剂盒(北京迈基诺基因科技股份有限公司)捕获上述目标基因文库,然后利用新一代测序仪HiSeq2500(Illumina公司,美国)进行高通量测序,得出数据后进行基因序列的生物信息学分析,找出致病基因并判断其突变性质。
1.3 Sanger测序验证根据二代测序检测结果,对患儿及其父母DNA标本AKR1D1突变基因进行Sanger测序验证。所用引物(表 2)根据AKR1D1基因的DNA序列使用Primer Premier 5.0软件设计,由北京迈基诺基因科技股份有限公司合成。聚合酶链反应体系为:2×Goldstar Buffer Mix 10 μL(北京康为世纪生物科技有限公司),上、下游引物各1 μL,DNA 1 μL,最后灭菌双蒸水7 μL至总体积为20 μL。反应条件为:95℃预变性10 min;随后分为4步,即第1步94℃变性30 s,64℃退火30 s,72℃延伸45 s,共3个循环;第2步94℃变性30 s,62℃退火30 s,72℃延伸45 s,共5个循环;第3步94℃变性30 s,60℃退火30 s,72℃延伸45 s,共10个循环;第4步94℃变性30 s,58℃退火30 s,72℃延伸45 s,共17个循环;最后再72℃延伸5 min。
| 表 2 AKR1D1基因突变位点的Sanger测序引物 |
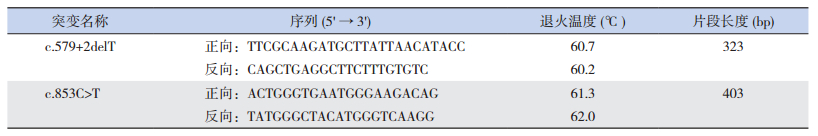
聚合酶链反应产物经琼脂糖凝胶电泳切胶纯化,由北京迈基诺基因科技股份有限公司测序。应用Chromas软件对测序结果进行分析,并应用DNAMAN软件与AKR1D1基因组参考序列进行比对(Ensemble Genome Browser: ENST00000122787)。突变的命名根据相关文献[14-15]。
1.4 生物信息学分析采用Human Splicing Finder(HSF, URL:http://www.umd.be/HSF3/)预测基因变异是否影响剪接,同时应用SWISS-MODEL软件(http://swissmodel.expasy.org/)分析变异前后AKR1D1蛋白质三级结构变化,推测基因变异影响蛋白质功能的可能机制。
本研究经暨南大学附属第一医院医学伦理委员会批准,并获得患儿父母签署书面知情同意书。
2 结果 2.1 遗传学分析结果代谢性肝病组套分析结果显示:患儿AKR1D1基因检测到2个突变,其中c.579+2delT为第5内含子的5' 端经典剪接位点的缺失突变,而c.853C > T(p.Q285X)为外显子7的无义突变(图 2)。Sanger测序验证证实,患儿为上述突变的复合杂合子,而其母亲和父亲分别为相应突变携带者(图 3)。

|
图 2 患儿AKR1D1基因代谢性肝病组套分析结果 上图为c.579+2delT突变,红框内可见AKR1D1基因的编码区第579+2位的野生型胸腺嘧啶T部分缺失;下图为c.853C > T(p.Q285X)突变,红框内可见AKR1D1基因的编码区第853位的野生型胞嘧啶C部分突变为胸腺嘧啶T。 |

|
图 3 患儿及其父母AKR1D1基因Sanger测序验证结果 患儿为c.579+2delT突变和c.853C > T(p.Q285X)突变的复合杂合子;父亲为c.853C > T(p.Q285X)突变携带者;母亲为c.579+2delT(Splicing)突变携带者。 |
查阅知网、万方、维普、美国Pubmed和人类基因突变数据库(human gene mutation database, HGMD),均未见AKR1D1基因c.579+2delT和c.853C > T(p.Q285X)突变的报道。HSF预测结果提示,c.579+2delT突变对野生型供体位点具有强破坏性,将产生新的剪接受体位点(野生型CV值为20.94,突变型CV值为71.9,ΔCV值为+243.36%),从而影响前体mRNA剪接,阻碍AKR1D1蛋白(Δ4-3-氧固醇5β-还原酶)的正常合成。SWISS-MODEL预测并构建野生型和突变型AKR1D1蛋白的三维结构,结果显示,突变c.853C > T导致AKR1D1蛋白翻译提前终止,致使突变型AKR1D1蛋白的三维结构缺失1个α-螺旋(图 4)。

|
图 4 AKR1D1蛋白质三维结构模型 A:显示野生型Δ4-3-氧固醇5β-还原酶,B:显示c.853C > T(p.Q285X)突变发生后的Δ4-3-氧固醇5β-还原酶。与野生型Δ4-3-氧固醇5β-还原酶(A)相比,突变型(B)C端缺失了一个α-螺旋(红框中紫色部分)。 |
2.2 治疗与结局
因病程中总胆汁酸水平基本正常,患儿以“1.先天性胆汁酸合成障碍?2.维生素D缺乏症;3.锌缺乏症”收住儿科。根据患儿临床特点及基因分析结果,患儿胆汁淤积病因确定为CBAS2,予口服鹅脱氧胆酸,同时给予康普力星补锌,并补充罗盖全、维生素AD和E等脂溶性维生素治疗半个月,患儿肝肿大和肝功能逐渐改善后出院。出院后继续以上治疗,3个月门诊复查,体重7.8 kg( < -2SD),身长69.5 cm( < -2SD),头围44.5 cm( > -2SD),黄疸完全消退,肝脏右肋下2.5 cm可及,质地软,且肝功能指标基本恢复正常。
3 讨论1988年Setchell等[16]首次报道了2例Δ4-3-氧固醇5β-还原酶缺陷病患儿。2003年,Lemonde等[5]提出AKR1D1基因突变为Δ4-3-氧固醇5β-还原酶缺陷病的病因,并将此病命名为CBAS2。研究发现酪氨酸血症1型[17]、新生儿血色沉着病[7, 18-19]和乙型肝炎[5]等疾病可继发Δ4-3-氧固醇5β-还原酶缺陷,与CBAS2鉴别困难。目前认为基因检测可用于确诊CBAS2,亦是鉴别CBAS2与继发性Δ4-3-氧固醇5β-还原酶缺陷病的唯一方法[7, 20]。本例患儿其主要临床表现为胆汁淤积性黄疸,伴有生长发育迟缓。多次生化检查发现血清转氨酶升高,总胆红素升高,且以结合胆红素为主,但γ-谷氨酰转肽酶和总胆汁酸基本正常。肝脏组织学检查示,肝小叶结构絮乱,肝细胞胞质呈细颗粒样或“毛玻璃”状,可见多核巨细胞,肝细胞内明显淤胆,间质和汇管区有大量淋巴细胞浸润,纤维组织和小胆管轻度增生。上述表现为典型CBAS2临床特点[1],但仅根据临床特点仍无法明确具体病因。本研究行代谢性肝病组套分析结合Sanger测序验证后,发现患儿为AKR1D1基因突变的c.579+2delT及c.853C > T(p.Q285X)复合杂合子,前者为来自母亲的剪接位点突变,后者为父源性无义突变,为CBAS2确诊提供了实验依据。
Δ4-3-氧固醇5β-还原酶拥有一个具有催化活性的核心结构域,此结构域中包括NAD(P)(H)结合区、活性区和底物结合位点[21]。该酶在NADPH的辅助下,在核心结构域进行双键还原反应,负责催化C-4/C-5双键结合在Δ4-3-酮类固醇上形成A/B环交接点,使平面类固醇产生A/B cis环,最终使胆汁酸具有兼性亲水亲脂特征,能够发挥其乳化脂肪和胆固醇的生理功能[4, 21-22]。本研究患儿为AKR1D1基因无义突变c.853C > T导致AKR1D1蛋白截短,其三级结构缺失1个α-螺旋,致使该蛋白核心结构域中的部分NAD(P)(H)结合区受损,最终影响酶蛋白活性;而突变c.579+2delT为新的剪接位点突变,经HSF软件预测,可导致mRNA影响剪接异常,产生异常蛋白,从而阻碍AKR1D1蛋白的正常合成。
CBAS2患者发病的关键病理生理环节是Δ4-3-氧固醇5β-还原酶的上游胆汁酸中间代谢物蓄积,而下游正常胆汁酸产物合成不足。口服初级胆汁酸,包括胆酸和鹅去氧胆酸,不仅可以提供人体必需的初级胆汁酸,而且可通过负反馈作用抑制胆固醇7α-羟化酶的表达,从而抑制异常胆汁酸的合成,减少肝毒性胆汁酸中间代谢物的产生,故本病采用初级胆汁酸替代治疗,可改善肝功能,避免肝脏进一步损害[8, 23]。肝移植是经药物治疗无法控制病情患儿的唯一选择,目前为止,仅有1例患儿进行了肝移植,术后2年10个月随访,预后良好[5]。本例患儿口服鹅去氧胆酸后肝肿大和肝功能进行性改善,暂无肝脏移植指征,但其远期预后有待随访观察。
本文系统性分析了1例CBAS2患儿的临床症状、体征和实验室检查特点,确诊患儿为AKR1D1基因新突变导致的CBAS2,并通过鹅去氧胆酸治疗取得良好效果。本研究扩展了AKR1D1基因突变谱,并为患者分子诊断、家系遗传咨询及产前诊断提供了依据。
| [1] | Clayton PT. Disorders of bile acid synthesis[J]. J Inherit Metab Dis, 2011, 34(3): 593–604. DOI:10.1007/s10545-010-9259-3 |
| [2] | Kondo KH, Kai MH, Setoguchi Y, et al. Cloning and expression of cDNA of human delta 4-3-oxosteroid 5 beta-reductase and substrate specifcity of the expressed enzyme[J]. Eur J Biochem, 1994, 219(1-2): 357–363. DOI:10.1111/ejb.1994.219.issue-1-2 |
| [3] | Charbonneau A, The VL. Genomic organization of a human 5beta-reductase and its pseudogene and substrate selectivity of the expressed enzyme[J]. Biochim Biophys Acta, 2001, 1517(2): 228–235. DOI:10.1016/S0167-4781(00)00278-5 |
| [4] | Di Costanzo L, Drury JE, Christianson DW, et al. Structure and catalytic mechanism of human steroid 5beta-reductase (AKR1D1)[J]. Mol Cell Endocrinol, 2009, 301(1-2): 191–198. DOI:10.1016/j.mce.2008.09.013 |
| [5] | Lemonde HA, Custard EJ, Bouquet J, et al. Mutations in SRD5B1(AKR1D1), the gene encoding delta(4)-3-oxosteroid 5beta-reductase, in hepatitis and liver failure in infancy[J]. Gut, 2003, 52(10): 1494–1499. DOI:10.1136/gut.52.10.1494 |
| [6] | Gonzales E, Cresteil D, Baussan C, et al. SRD5B1(AKR1D1) gene analysis in delta(4)-3-oxosteroid 5beta-reductase deficiency:evidence for primary genetic defect[J]. J Hepatol, 2004, 40(4): 716–718. |
| [7] | Ueki I, Kimura A, Chen HL, et al. SRD5B1 gene analysis needed for the accurate diagnosis of primary 3-oxo-Delta4-steroid 5beta-reductase defciency[J]. J Gastroenterol Hepatol, 2009, 24(5): 776–785. DOI:10.1111/jgh.2009.24.issue-5 |
| [8] | Seki Y, Mizuochi T, Kimura A, et al. Two neonatal cholestasis patients with mutations in the SRD5B1(AKR1D1) gene:diagnosis and bile acid profiles during chenodeoxycholic acid treatment[J]. J Inherit Metab Dis, 2013, 36(3): 565–573. DOI:10.1007/s10545-012-9526-6 |
| [9] | Morgan NV, Hartley JL, Setchell KD, et al. A combination of mutations in AKR1D1 and SKIV2L in a family with severe infantile liver disease[J]. Orphanet J Rare Dis, 2013, 8: 74. DOI:10.1186/1750-1172-8-74 |
| [10] | Zhao J, Fang LJ, Setchell KD, et al. Primary Δ4-3-oxosteroid 5β-reductase deficiency:two cases in China[J]. World J Gastroenterol, 2012, 18(47): 7113–7117. DOI:10.3748/wjg.v18.i47.7113 |
| [11] | 方玲娟, 王建设. 先天性胆汁酸合成障碍与胆汁淤积性肝病[J]. 临床肝胆病杂志, 2010, 26(6): 585–588. |
| [12] | 代东伶. 先天性胆汁酸合成障碍[J]. 临床儿科杂志, 2015, 33(4): 301–305. |
| [13] | 胡长霞, 黄志华. 先天性胆汁酸合成障碍的诊治[J]. 中国临床医生, 2012, 40(11): 13–15. DOI:10.3969/j.issn.1008-1089.2012.11.005 |
| [14] | den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations:a discussion[J]. Hum Mutat, 2000, 15(1): 7–12. DOI:10.1002/(SICI)1098-1004(200001)15:1<>1.0.CO;2-B |
| [15] | den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations[J]. Hum Genet, 2001, 109(1): 121–124. DOI:10.1007/s004390100505 |
| [16] | Setchell KD, Suchy FJ, Welsh MB, et al. Delta 4-3-oxosteroid 5 beta-reductase deficiency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis[J]. J Clin Invest, 1988, 82(6): 2148–2157. DOI:10.1172/JCI113837 |
| [17] | Clayton PT, Patel E, Lawson AM, et al. 3-oxo-delta 4 bile acids in liver disease[J]. Lancet, 1988, 1(8597): 1283–1284. |
| [18] | Shneider BL, Setchell KD, Whitington PF, et al. Delta 4-3-oxosteroid 5 beta-reductase deficiency causing neonatal liver failure and hemochromatosis[J]. J Pediatr, 1994, 124(2): 234–238. DOI:10.1016/S0022-3476(94)70310-8 |
| [19] | Siafakas CG, Jonas MM, Perez-Atayde AR. Abnormal bile acid metabolism and neonatal hemochromatosis:a subset with poor prognosis[J]. J Pediatr Gastroenterol Nutr, 1997, 25(3): 321–326. DOI:10.1097/00005176-199709000-00015 |
| [20] | Yanagi T, Mizuochi T, Homma K, et al. Distinguishing primary from secondary Δ(4)-3-oxosteroid 5β-reductase (SRD5B1, AKR1D1) deficiency by urinary steroid analysis[J]. Clin Endocrinol (Oxf), 2015, 82(3): 346–351. DOI:10.1111/cen.2015.82.issue-3 |
| [21] | Russell DW, Setchell KD. Bile acid biosynthesis[J]. Biochemistry, 1992, 31(20): 4737–4749. DOI:10.1021/bi00135a001 |
| [22] | Rižner TL, Penning TM. Role of aldo-keto reductase family 1(AKR1) enzymes in human steroid metabolism[J]. Steroids, 2014, 79: 49–63. DOI:10.1016/j.steroids.2013.10.012 |
| [23] | Gonzales E, Gerhardt MF, Fabre M, et al. Oral cholic acid for hereditary defects of primary bile acid synthesis:a safe and effective long-term therapy[J]. Gastroenterology, 2009, 137(4): 1310–1320.e1-3. DOI:10.1053/j.gastro.2009.07.043 |