 2017, Vol. 19
2017, Vol. 19



患者,女,汉族,因生后气促10 min入院。第2胎第1产,剖宫产出生,出生胎龄29+2周、体重1 210 g,1 min Apgar评分8分,5 min Apgar评分9分。胎膜早破近4天,羊水清亮,量约200 mL。患儿母亲29岁,孕中晚期发现羊水过多,产前唐氏筛查低风险,未行羊水穿刺。父亲29岁,体健。否认家族中遗传性疾病史。
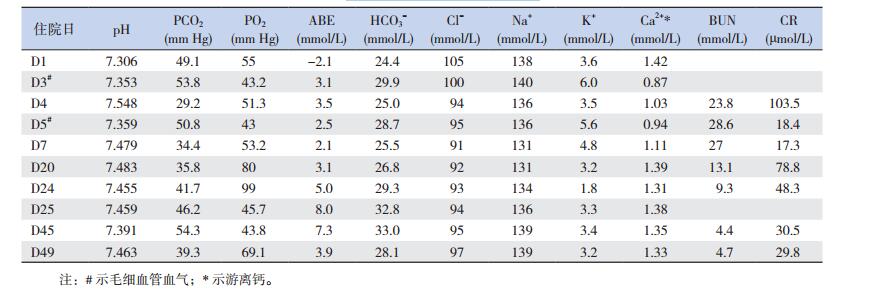
入院体查:体温36.5℃,脉搏140次/min,呼吸60次/min,血压81/46 mm Hg,身长41 cm,体重1 210 g,头围28 cm。头颅五官无畸形,早产儿貌,胸廓略饱满,轻度吸气性三凹征,两肺呼吸音低,未闻及罗音,心音有力,腹软,肝、脾无肿大,四肢肌张力偏低。实验室检查:血、尿常规及肝功能正常,CRP、血培养、甲状腺功能无异常。入院第1周(糖速为每分钟4 mg/kg)微量血糖7.1~10.5 mmol/L,尿糖阴性;第2周(糖速每分钟5~6 mg/kg)血糖逐渐下降至正常。入院第4天的尿素氮23.8 mmol/L(参考值:1.1~9.1 mmol/L)、肌酐正常;此后尿素氮均 > 20 mmol/L,肌酐正常,第24天尿素氮、肌酐正常。入院第4天血气分析提示低氯性代谢性碱中毒(pH 7.548, HCO3-25.0 mmol/L,PCO2 29.2 mm Hg,ABE 3.5 mmol/L,Cl-94 mmol/L,K+ 3.5 mmol/L);此后多次血气均提示低氯性碱中毒,伴低钾、低钠血症,血镁正常。见表 1。尿比重正常。血串联质谱及尿气相质谱分析无异常。腹部平片未提示异常。肾脏B超未见异常。心脏B超提示动脉导管未闭及卵圆孔未闭。
| 表 1 住院期间主要生化及血气结果 |
温箱保暖(箱温35℃、湿度70%),并立即静脉营养:第1天补液量为80 mL/kg,每天增加15~20 mL/kg,第6天达160 mL/kg,尿量为每小时4.0~5.9 mL/kg;第7天体重降至990 g(降低18.1%),第19天回升至出生体重。入院24 h内开始鼻饲,至第27天全胃肠营养。曾予肺表面活性物质治疗,持续呼气末正压通气11 d,高流量正压通气37 d。
2 诊断思维患儿入院后首先表现出三个问题:血糖增高、氮质血症及体重不增。新生儿高血糖需要注意感染、应激、糖调节功能不成熟及新生儿糖尿病等。患儿虽有胎膜早破,但无感染中毒症状,动态监测血常规及CRP均正常,血培养阴性,无感染依据。患儿无窒息史,无寒冷损伤,不支持应激所致血糖增高。早产儿糖调节功能不成熟常与早产儿胰岛β细胞功能不成熟有关,多为暂时性,本例患儿为早产,血糖轻度增高,不排除此种可能性。新生儿糖尿病发病率低,可能性较小,进一步监测血糖,必要时查血浆胰岛素、C肽、胰腺CT等。而新生儿氮质血症常见原因有蛋白摄入不足/过量、肾功能不全或血容量不足。患儿生后立即静脉营养,每日氨基酸在2~3 g/kg,符合早产儿生理需求,血白蛋白也正常,不支持蛋白摄入不足。患儿无高钾血症、血肌酐正常、肾小球滤过率正常,可排除肾功能不全。患儿为早产儿,肾脏浓缩功能差,尿量多,而且有呕吐,存在血容量不足导致氮质血症可能。患儿体重不增可能与糖摄入限制导致的热量不足,以及尿量增多、呕吐导致的液体量相对不足有关。另外,还需注意少见的遗传缺陷所致的蛋白质、糖类、脂肪酸等主要营养物质代谢障碍,但串联质谱分析无异常。予增加补液、减少蛋白摄入,动态监测血糖、肾功能、电解质等,患儿血糖、尿素氮恢复正常,但体重并不随肠道喂养建立、热量供给增加而增加,且逐渐出现低钠、低钾、低氯性代谢性碱中毒。引起水电酸碱失衡的常见病因有尿崩症,盐皮质激素分泌异常引起的继发性肾小管功能异常如肾上腺皮质增生症、原发性醛固酮增生症等疾病,肾小管原发性疾病如肾小管性酸中毒、巴特综合征等。尿崩症为抗利尿激素分泌不足或失活所致,特点是低渗性多尿、高钠血症,新生儿期发病较少。本例新生儿期发病,尿比重正常,血钠降低,不支持尿崩症。先天性肾上腺皮质增生症可有体重不增、脱水症状以及高钾、低钠性酸中毒,本例患儿表现为低钠、低钾、低氯性代谢性碱中毒、17-α羟孕酮正常,与先天性肾上腺皮质增生症不符。原发性醛固酮增多症表现为顽固性高血压、低血钾,而大量失钾导致肾小管上皮细胞浓缩功能减退,出现多尿症状,也可出现轻度代谢性碱中毒表现,症状与本例患儿较相似,但本例患儿无原发性醛固酮增多症典型的高血压表现。常见的原发性肾小管疾病如肾小管性酸中毒可有多尿、脱水、体重不增,但其突出特点为肾小管排H+障碍(Ⅰ型)或肾小管对HCO3-重吸收障碍(Ⅱ型)以及代谢性酸中毒,本例患儿为代谢性碱中毒。少见的肾小管疾病如巴特综合征,可表现为多尿,低氯性代谢性碱中毒合并低钾血症、低钠血症,可解释本例的低钠、低钾、低氯性代谢性碱中毒。综合以上分析,不能排除原发性醛固酮增多症及巴特综合征,进一步查ACTH、醛固酮、血管紧张素、肾素等相关激素水平,并根据其结果予以基因筛查。
3 进一步检查醛固酮1 062 pg/mL(参考值:10~160 pg/mL)。肾素1 538 pg/mL(参考值:4~24 pg/mL)。血管紧张素Ⅰ正常;血管紧张素Ⅱ 1 145.7 pg/mL(参考值:16.2~64.2 pg/mL)。
基因检测发现SLC12A1基因第11号外显子c.1435C > G杂合突变和第12号外显子c.1478_1478delG杂合缺失突变。见图 1。

|
图 1 患儿基因突变检出情况 SLC12A1基因第11号外显子c.1435C>G杂合突变和第12号外显子c.1478_1478delG杂合缺失突变,突变位点如箭头所示。 |
4 诊断及确诊依据
患儿确诊为新生儿巴特综合征。依据:(1)患儿母亲孕中晚期羊水过多;(2)患儿多尿、脱水,体重不增,反复呕吐;(3)持续低氯性代谢性碱中毒,低钾、低钠血症,醛固酮、肾素、血管紧张素Ⅱ均升高;(4)SLC12A1基因突变。
5 临床经过诊断明确后以予补钠、补钾等对症治疗。出院后继续口服氯化钠、氯化钾。84天龄复查,pH 7.53(参考值:7.35~7.45),HCO3-30 mmol/L(参考值:22~27 mmol/L),PCO2正常,ABE 7 mmol/L(参考值:-3~3mmol/L),Cl-、K+、Na+正常。至生后8个月,神经精神发育水平与纠正月龄基本相符,定期复查血气仍有轻度代谢性碱中毒,电解质基本正常。
6 讨论巴特综合征(Bartter syndrome, BS)是一种常染色体遗传性肾小管疾病,1962年由Federic Bartter首次报道[1]。目前认为其主要发病机制为氯化物在髓袢升支粗段(thick ascending limb of the loop of Henle, TAL)的再吸收障碍,导致低钾血症、代谢性碱中毒、肾素-血管紧张素-醛固酮系统激活,而血压正常或偏低可能与肾脏钠的丢失及肾小球旁器增生有关[2-3]。多数学者将巴特综合征分为新生儿型巴特综合征(neonatal Bartter syndrome, nBS)、经典型巴特综合征(classical Bartter syndrome, cBS)、巴特综合征变异型(Gitelman syndrome, GS)三种。根据基因突变位点分型则分为6种,即BSⅠ~Ⅴ型和GS型,其中BSⅤ型为常染色体显性遗传,其余均为常染色体隐性遗传[3-4]。国外文献分型通常不包含GS [2, 5]。
新生儿BS最早的异常表现出现于宫内,包括胎儿多尿、宫内生长受限等,孕母24~30周羊水过多,羊水检查常提示钠、钾正常,而氯升高[2, 10]。对BS宫内表现的认识对于BS的早期诊断具有很重要的意义。新生儿BS发病率据国外统计为1/5~10万[6],国内报道较少,且基本都在生后至少数月方确诊。吴文燊等[7]报道1例出生胎龄29周的新生儿巴特综合征,表现为多尿、体重不增、代谢性碱中毒及低钾血症,与本文患儿类似。国外近年来也有类似的早产儿巴特综合征的报道[8-9]。
巴特综合征的发病机制较复杂,主要认为是基因缺陷导致了TAL中NKCC2、ROMK、CLC-Kb等转运体或通道蛋白缺陷,使NaCl在TAL的重吸收减少,到达远端肾组织的NaCl增多,钠、钾交换后钾排泄增加,随后肾素-血管紧张素-醛固酮系统激活,从而出现多尿、失盐、容量不足,并导致低氯性代谢性碱中毒、低钾血症、低钠血症、高尿钙等一系列症状,但由于BS患者高前列腺素水平及缓激肽释放酶活性增高等因素,血压基本正常。BS I型是由于Na+-K+-2Cl-协同转运体NKCC2缺陷所致,导致K+丢失增加。而NKCC2由常染色体15q15的SLCl2A1基因所编码。BSⅡ型由ROMK缺陷所致,可导致TAL的重吸收功能紊乱。ROMK由位于1lq24的KCNJl基因编码,是ATP依赖的钾通道蛋白。BSⅢ型是由位于lp36的CLCNKB基因突变所致,主要影响Cl-的重吸收。BS IV型是nBS的一种亚型,是1p31的BSND基因突变所致,此序列编码的蛋白质为肾特异性氯通道的β亚基,最终导致Cl-重吸收减少。BSⅤ型由位于3q13.3-q21的基因CASR突变所致,最终ROMK活性受到抑制,从而NKCC2运转异常[3]。本例患儿为早产,母亲孕中晚期发现羊水过多,新生儿尿量增多,存在低氯、低钾性代谢性碱中毒,肾素、血管紧张素、醛固酮升高,但无高血压发生,SLC12A1基因错义突变,符合BSⅠ型特点。
BS的治疗根据基因型的不同有所不同,原则均为减少钾的丢失,减少尿量。如发现母孕期羊水持续增加,需要注意新生儿巴特综合征可能,胎儿尿液分析发现氯化物增加有助于诊断[10]。在确诊后、娩出前,孕母可予以吲哚美辛治疗,但需要注意吲哚美辛可能引起的胎儿动脉导管收缩,对胎儿动脉导管开放情况应予监测[2]。对于BS患儿,纠正脱水及电解质紊乱是两个重要方面。前列腺素酶抑制剂对于NKCC2转运体及ROMK通道出现基因突变者效果好;吲哚美辛及阿司匹林、布洛芬等药物均可有效减少尿量,但需要注意肠穿孔等副作用,对于早产儿尤需慎重[6]。另外,保钾利尿剂可能改善低钾血症,但作用短暂,且可导致水、钠丢失及血容量降低,需要谨慎对待。
nBS患者神经、精神发育通常是正常的,生长迟缓几乎发生在所有患者;电解质紊乱可导致慢性肾小管间质性肾病及肾小球滤过率的退化,但需透析或肾移植的肾功能衰竭发生较罕见。未经治疗的新生儿BS可能死于脱水、电解质紊乱、感染,及时合理的治疗可以改善临床症状以及生长发育[2, 11]。
7 结语BS为新生儿期少见的一种临床疾病,起病隐匿,早期诊断困难。本例患儿出生胎龄29周,生后早期即出现多尿,体重不增,呕吐、腹部膨隆,诊断分析曾局限在早产儿肾脏浓缩功能不全以及液体量不足等常见原因,直至出现电解质及酸碱平衡紊乱方考虑肾小管疾病,继而发现醛固酮、肾素、血管紧张素升高以及SLC12A1基因突变,得以确诊nBS。因此,对于难以解释的多尿、电解质紊乱,尿电解质分析及综合评价肾小管功能非常重要;母孕期羊水过多的需要警惕nBS,胎儿尿液分析有助诊断。
| [1] | Batter FC, Pronove P, Gill JR Jr, et al. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome[J]. Am J Med, 1962, 33: 811–828. DOI:10.1016/0002-9343(62)90214-0 |
| [2] | Bhat YR, Vinayaka G, Sreelakshmi K. Antenatal bartter syndrome:a review[J]. Int J Pediatr, 2012, 2012: 857136. |
| [3] | 周美央, 梁华. 巴特综合征研究进展[J]. 国际泌尿系统杂志, 2007, 27(1): 124–129. |
| [4] | Fremont OT, Chan JC. Understanding Bartter syndrome and Gitelman syndrome[J]. World J Pediatr, 2012, 8(1): 25–30. DOI:10.1007/s12519-012-0333-9 |
| [5] | Brochard K, Boyer O, Blanchard A, et al. Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome[J]. Nephrol Dial Transplant, 2009, 24(5): 1455–1464. DOI:10.1093/ndt/gfn689 |
| [6] | 曹力, 陈朝英, 陈大坤, 等. 新生儿型巴特综合征1例及文献复习[J]. 中国实用儿科杂志, 2003, 18(1): 36–39. |
| [7] | 吴文燊, 叶秀桢, 王艳丽, 等. 新生儿型Bartter综合征1例[J]. 中国新生儿科杂志, 2014, 29(6): 418–419. |
| [8] | Wong AC, Chan LG. Neonatal Bartter syndrome[J]. Med J Malaysia, 2014, 69(5): 229–230. |
| [9] | Flores FX, Ojeda FJ, Calhoun DA. Bartter syndrome:presentation in an extremely premature neonate[J]. J Perinatol, 2013, 33(8): 661–662. DOI:10.1038/jp.2013.13 |
| [10] | Rachid ML, Dreux S, Czerkiewicz I, et al. Fetal urine biochemistry in antenatal Bartter syndrome:a case report[J]. Clin Case Rep, 2016, 4(9): 876–878. DOI:10.1002/ccr3.471 |
| [11] | Puricelli E, Bettinelli A, Borsa N, et al. Long-term follow-up of patients with Bartter syndrome type I and Ⅱ[J]. Nephrol Dial Transplant, 2010, 25(9): 2976–2981. DOI:10.1093/ndt/gfq119 |