 2017, Vol. 19
2017, Vol. 19



婴儿肝衰竭综合征1型(infantile liver failure syndrome type 1, ILFS1, OMIM #615438)是一种由胞质亮氨酰-tRNA合成酶基因(leucyl-tRNA synthetase gene, LARS)突变导致的常染色体隐性遗传病[1-2]。LARS基因定位于染色体5q32,含有32个外显子,转录子全长4 766 bp,编码由1 176个氨基酸组成并位于胞质的亮氨酰-tRNA合成酶(leucyl-tRNA synthetase, LeuRS)[3]。LeuRS属于第一类氨基酰-tRNA合成酶(aaRS),其作用是通过氨酰化反应把氨基酸共价连接到与其对应的tRNA上,而氨酰化反应的第一步是氨基酸的活化反应,即ATP和氨基酸发生反应生成氨基酰-AMP及焦磷酸;第二步是tRNA的氨酰化反应,即氨基酰被转移到tRNA的3'端核糖上,产生氨基酰-tRNA,并释放AMP。此外,aaRS还具有水解错误活化的氨基酰-AMP(转移前编辑,pre-transfer editing)和错配的氨基酰-tRNAs(转移后编辑,post-transfer editing)的功能,确保从mRNA到蛋白质翻译过程的高度精确性[4-7]。
2012年,Casey等[1]首次报道ILFS1,描述了一个爱尔兰家系的6位本病患者。之后该课题组又陆续发现4位ILFS1患者[2],其中3位来自于另一爱尔兰家庭,还有一位来自美国的犹太人。迄今未见本病非白人患者的报道。ILFS1临床表现常涉及多个系统,患者通常在1岁以内表现为低出生体重、肝功能异常、低蛋白血症、贫血和生长发育迟缓等,肝脏病理显示显著的脂肪肝和纤维化。肝功能异常主要出现在发热时,且病情随着年龄增长逐渐改善。部分患者可能出现肝功能衰竭、惊厥和脑病等严重表现,甚至死亡。本研究旨在报道首例非白人ILFS1患者的临床特点和分子诊断结果,为本病诊治研究提供参考。
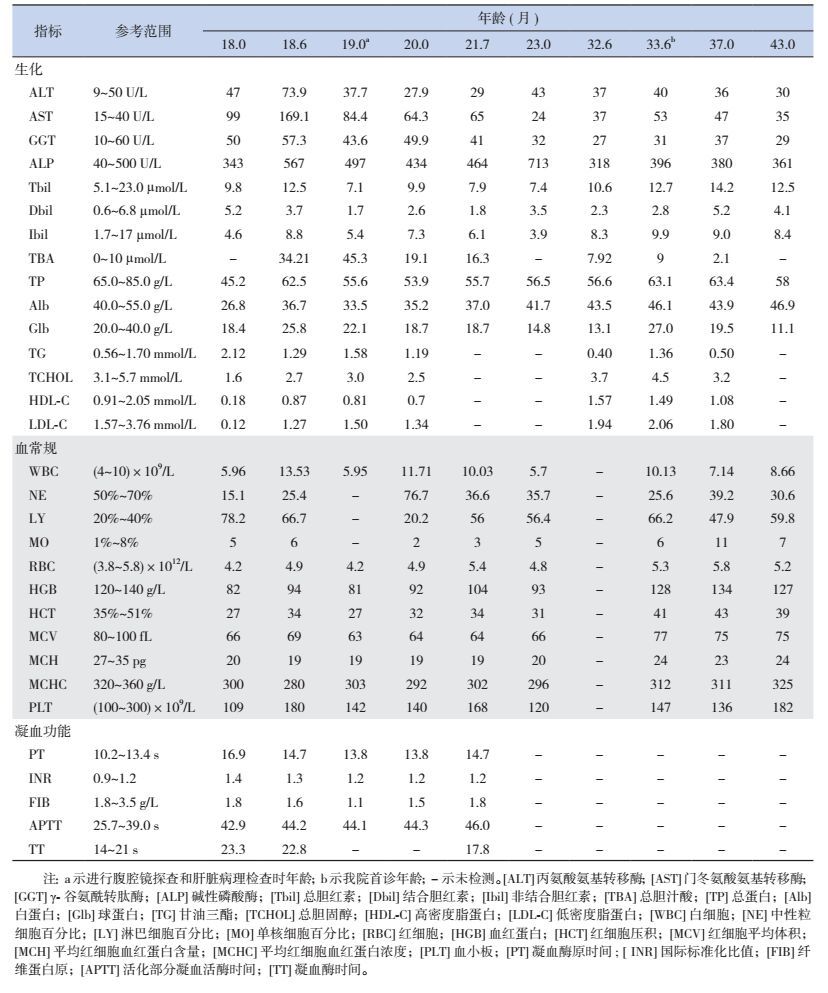
1 资料与方法 1.1 研究对象患者,男,2岁9个月,因发现肝脾肿大1年余入院。患者1岁5个月时因发热2天于当地医院就诊,血常规示中度贫血,B超示肝脾肿大(具体不详)。诊断“肝脾肿大待查、急性上呼吸道感染、中度贫血”。抗感染1天后热退,补铁、护肝治疗半个月后贫血及肝脾肿大无改善。1岁6个月查体示肝右肋下8 cm、剑突下5 cm,质硬;脾肋下3 cm,质中;实验室检查发现丙氨酸氨基转移酶和门冬氨酸氨基转移酶偏高、低蛋白血症、小细胞低色素性贫血和凝血功能异常等,见表 1;骨髓细胞学检查未见明显异常,巨细胞病毒、EB病毒、乙肝和丙肝等病毒及自身免疫性肝炎相关抗体检查阴性;血串联质谱遗传代谢病筛查示多种氨基酸轻度下降,尿有机酸气相质谱分析提示酮尿;SLC25A13基因c.851_854del4、c.1638_1660dup、IVS6+5G > A、IVS16ins3kb和IVS11+1G > A突变筛查均阴性。肝脾肿大、贫血原因不明,予护肝等对症等治疗,临床表现无明显改善。1岁7个月曾行B超及CT检查,提示肝内血管瘤可能性大;行腹腔镜探查和肝活检术,发现肝脏明显肿大,表面布满大小不等黄白色结节;肝脏病理显示肝硬化和脂肪肝。2岁8个月时行代谢性肝病相关的544个基因外显子高通量测序,在SLC25A13基因中发现一父源性的杂合突变c.1661G > A(p.R554Q),其余基因未见明确致病突变。患者肝功能及贫血症状随年龄增长渐改善,但病因仍不明。
| 表 1 患儿历次生化、血常规及凝血功能检查结果 |
患儿系第一胎第一产,足月顺产,出生体重2 500 g(-2.1 SD),身长50 cm(-0.23 SD)。生后3d出现皮肤轻度黄染,持续1个月,停止母乳喂养后黄疸渐消退。平素易患上呼吸道感染,9月龄时曾患重症肺炎。否认家族类似病史。
体格检查:体重13.5 kg(-0.04 SD),身长89 cm(-1.23 SD),头围47.5 cm(-1.25 SD)。神清,皮肤巩膜无黄染,双肺呼吸音清,心界不大,心律齐,心音有力,各瓣膜区未闻及杂音。腹平软,肝脏右肋下2.5 cm、质中,脾未触及。四肢肌张力正常,腹壁和膝腱反射可引出,克布氏征、巴氏征均阴性。
辅助检查:血常规和生化检查大致正常(表 1)。肝脏病理切片会诊(图 1)提示:肝内大小不等的结节形成,纤维瘢痕及纤维间隔内较大量炎细胞浸润,小胆管增生明显,轻度界面炎;部分结节内肝细胞大泡性脂肪变性,窦周纤维化可见,考虑肝硬化(活动期)。

|
图 1 肝脏组织病理检查结果 A(HE染色,100×):肝内大小不等的结节形成,纤维瘢痕及纤维间隔内大量炎细胞浸润,轻度界面炎,部分结节内肝细胞大泡性脂肪变性;B(Masson染色,100×):纤维组织增生分隔肝实质(胶原纤维绿色,肌纤维红色);C(CK19染色,200×):小胆管增生明显(棕色,如箭头所示);D(Vimentin染色,200×):窦周纤维化(棕色,如箭头所示)。 |
1.2 SLC25A13基因高频突变筛查和Sanger测序分析
本研究获得暨南大学附属第一医院医学伦理委员会批准及患儿父母知情同意。采集患儿及其父母静脉血2 mL(EDTA抗凝),按DNA抽提试剂盒(Simgen公司,中国)说明书提取基因组DNA。利用PCR、PCR-RFLP和LA-PCR技术筛查SLC25A13基因c.851_854del4、c.1638_1660dup、IVS6+5G > A和IVS16ins3kb 4种高频突变[8-10]。利用PCR和LA-PCR技术扩增SLC25A13基因18个外显子及其侧翼区[9-13],将产物纯化后,通过ABI 3730xl测序仪(Invitrogen,美国)及应用双脱氧链终止法技术对样品进行Sanger测序。
1.3 SLC25A13 cDNA克隆分析参考文献[14]进行SLC25A13 cDNA克隆分析。用淋巴细胞分离液(MP Biomedicals,美国)分离外周血淋巴细胞,加RNAiso plus(TaKaRa, 日本)裂解细胞后,提取总RNA。以总RNA为模板,按照M-MLV逆转录酶(Invitrogen, 美国)说明书合成第一链cDNA。巢式PCR扩增SLC25A13基因编码区,切胶纯化PCR产物后与PMD 18-T Vector(TaKaRa, 日本)进行连接克隆。转化感受态细胞,涂布于加有Amp(50~100 μg/mL)的LB平板培养基上,37℃培养箱培养过夜,形成单菌落。随机挑选单菌落,接种于含Amp的500 μL LB液体培养基中,37℃振荡培养3~5 h。取1 μL菌液为模板,进行PCR扩增,电泳检测有无目的条带,并挑选24个阳性克隆菌液进行Sanger测序。
1.4 靶向外显子组捕获测序分析提取基因组DNA 3~5 μg,将其打断、扩增,建立含有与代谢性肝病相关目标基因(JAG1、ATP8B1、GALT、MUT、MMAA、MARS和LARS等共249个基因)的全基因组文库,并对文库样本进行定量,保证文库样本总量在3 μg以上。用液相捕获试剂盒(MyGenostics, 中国)捕获目标基因,利用测序仪HiSeq 2000(Illumina, 美国)进行高通量测序,平均深度不小于200×。获取原始短序列,通过生物信息学分析单核苷酸多态性(single nucleotide polymorphism, SNP)或插入/缺失(Indel),找出相关基因的突变信息。靶向外显子组捕获测序分析由北京迈基诺基因科技有限责任公司完成。
1.5 Sanger测序验证根据外显子组捕获测序结果,对患儿及其父母DNA进行Sanger测序,验证LRAS基因突变。根据LRAS基因序列,运用Premier 5.0软件设计引物(上海立菲生物技术有限公司合成)。其中,外显子12的上游引物为F9_F(5'-AGTCCCACAGCGTAGTTCAGC-3'),下游引物为F9_R(5'-TCTTGGTGCATCACTTTCTGC-3'),产物为321 bp;而外显子21的上游引物为F8_F(5'-AAGTGATCCTCCCACCTTGG-3'),下游引物为F8_R(5'-TTTATGTAGCCAGTCCTTTGATTTG-3'),产物为402 bp。PCR反应体系包含:5 μL 10×Buffer,4 μL dNTP,0.25 μL rTaq DNA聚合酶(TakaRa, 日本),各1 μL上下游引物(10 μmol/L),0.1μg DNA,加灭菌双蒸水至50 μL;扩增条件为94℃预变性5 min,94℃变性30 s,53℃退火30 s,72℃延伸50 s,共35个循环,最后再72℃延伸7 min。PCR产物纯化后进行Sanger测序。
1.6 生物信息学分析突变的人群频率从大数据库Exom Aggregation Consortium(ExAC)(http://exac.broadinstitute.org/)中搜索。分析氨基酸保守性,不同物种LeuRS同源蛋白序列从NCBI(www.ncbi.nlm.nih.gov)数据库下载,采用软件Clustal Omega(http://www.ebi.ac.uk/Tools/msa/clustalo/)进行比对。突变致病性通过PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、PROVEAN(provean.jcvi.org/index.php)和MutationTaster(http://mutationtaster.org/MutationTaster/index.html)三种软件进行预测。此外,LeuRS蛋白的结构模型使用在线软件SWISS MODEL构建(http://swissmodel.expasy.org),并采用SWISS-PdbViewer 4.1.0(http://www.Expasy.org/spdbv/)对突变的蛋白结构进行预测。
2 结果 2.1 SLC25A13基因突变分析和cDNA克隆分析结果SLC25A13基因高频突变筛查和测序分析仅检测到一个父源性的突变c.1658G > A。SLC25A13 cDNA克隆分析结果显示,来自父源性的等位基因的克隆子共有7个,均含有点突变c.1658G > A;而来自母源性的等位基因的克隆子共有17个,但未发现异常的转录子,因此排除了希特林缺陷病导致的新生儿肝内胆汁淤积症。
2.2 外显子组捕获测序和Sanger测序结果代谢性肝病相关的249个基因外显子捕获测序检测到患儿LARS基因两个突变c.2133_2135del和c.1183G > A。Sanger测序结果(图 2)证实,患儿为LARS基因c.2133_2135del和c.1183G > A突变的复合杂合子,前者来自父亲,后者来自母亲。

|
图 2 患儿及其父母LARS基因的Sanger测序图 患者为c.1183G > A和c.2133_2135del突变的复合杂合子;其父亲和母亲分别为突变c.2133_2135del和c.1183G > A的携带者。突变位点如箭头所示。 |
2.3 生物信息学分析结果
突变c.1183G > A(p.D395N)和c.2133_2135del(p.L712del)在ExAC大数据库中的人群频率分别为0.005%和0.002%。比对包括人类、黑猩猩、狗、奶牛、大鼠、小鼠、鸡、热带爪蟾和线虫在内的9种真核生物物种之间的LeuRS同源蛋白序列,结果显示D395和L712均为保守氨基酸。两种突变的MutationTaster分析均 > 0.9999,PROVEAN分析的分数分别为-4.92和-13.23;PolyPhen-2分析错义突变c.1183G > A的分数为0.999,均提示此两种突变可能为致病性突变。
进行蛋白结构预测时,p.D395野生型选取的蛋白结构模型为人胞质的亮氨酰-tRNA合成酶的编校结构域(2WFD,258-509位氨基酸),序列一致性为99.21%[15]。采用SWISS-PdbViewer 4.1.0软件将p.D395野生型模型的第395位氨基酸由D变成N,突变后进行模型能量最小化,并比对两者构象的差异。第395位氨基酸由天冬氨酸变成天冬酰胺,使其附近氨基酸之间的氢键距离改变,同时伴有氢键生成和消失(图 3)。由于2WFD不能覆盖L712,分析突变p.L712del的野生型和突变型选取的蛋白结构模型为嗜热古菌亮氨酰-tRNA合成酶和tRNALeu复合物晶体结构(1wz2.1.B),序列一致性分别为32.41%和32.45%[16]。两者进行比对:第712位亮氨酸缺失,可破坏709-713和753-755位氨基酸构成的β折叠结构,同时,KMSKS模体(第716-720位氨基酸)与第155-164位氨基酸构成的α螺旋结构缠绕在一起(图 4)。

|
图 3 p.D395N突变前后空间结构预测图 第395位氨基酸由天冬氨酸(A、B)变成天冬酰胺(C、D),使其附近氨基酸之间的氢键距离改变,同时伴有氢键生成和消失;T298和D399与附近氨基酸之间的氢键距离也受影响。绿色虚线表示氢键,相应数字表示氢键距离。 |

|
图 4 p.L712del突变前后空间结构预测图 A、B为野生型蛋白结构模型,C、D为突变后蛋白结构模型,第712位亮氨酸缺失破坏了709-713和753-755位氨基酸构成的β折叠结构,而位于第716-720位氨基酸的KMSKS模体与第155-164位氨基酸构成的α螺旋结构缠绕在一起(如C图箭头所示)。 |
2.4 诊治经过与结局
根据患儿病史、临床表现和实验室检查,考虑遗传性肝病导致的肝硬化。行SLC25A13基因突变分析和cDNA克隆分析,排除希特林缺陷病。但病因仍不明,未予特殊治疗。最终(3岁时)经代谢性肝病相关基因外显子组捕获二代测序和Sanger测序验证,确诊为LARS基因突变导致的ILFS1。目前随访至4岁,肝功能正常,但仍有低蛋白血症,无贫血,见表 1;B超显示肝内回声较前改善。
3 讨论本研究患儿主要表现为肝脾肿大以及出生体重偏低、肝功能异常、低蛋白血症、贫血、凝血功能异常等,同时肝脏病理检查显示肝硬化和脂肪肝,这些表现均与希特林缺陷病的临床特点相似[17]。此外,SLC25A13基因检测还发现一杂合突变点c.1661G > A(p.R554Q),因此高度疑诊希特林缺陷病。然而外周血淋巴细胞SLC25A13 cDNA克隆分析在另一个等位基因上没有发现异常的转录子,从而排除了希特林缺陷病。提示希特林缺陷病的诊断需要借助SLC25A13基因突变分析或mRNA分析等特异性手段[17-18]。该患者骨髓细胞学检查、病原体检查及自身免疫性肝炎相关抗体检查均未见明显异常,腹腔镜探查和肝脏病理检查也排除了肿瘤可能;此外,血串联质谱遗传代谢病筛查提示多种氨基酸改变,因此高度怀疑遗传性肝病。但目前报道的相关基因多达数百个,根据临床和常规检测手段难以确定致病基因。本研究通过外显子组捕获二代测序及Sanger测序验证,确认该患儿为LARS基因c.2133_2135del和c.1183G > A突变的复合杂合子。这两个突变在ExAC大数据库中的人群频率非常低,且均位于重要功能区内,多种生物信息学软件从进化保守性、蛋白结构和功能、序列同源性等方面的分析均提示这两个突变可能致病。值得注意的是,患者在外院曾接受过高通量测序检查,由于检测目标没有覆盖LARS基因而漏诊,提示高通量测序的检测范围对于遗传性肝病的病因诊断至关重要。
LARS基因编码的人胞质LeuRS以多酶复合体(multi-tRNA synthetase complex, MSC)形式存在。根据特有的结构特征,LeuRS归属于第一类aaRS,其氨酰化反应的催化活性中心是一个由两个β3α2结构组成的Rossmann折叠结构域,首尾包含HIGH和KMSKS两个特征肽段,能与ATP结合[19]。在Rossmann折叠的两个β3α2结构之间插入一个连接肽段CP1结构域(connecting peptide 1 domain, 260-509aa),具有编辑功能[4, 20]。该编辑活性中心含有两个保守位点,一个是富苏氨酸区,其中T298高度保守,与底物专一性结合相关;另一个是天冬氨酸D399[21]。此外,LeuRS还含有一个接近C末端的结合反密码子的结构域和C末端附加结构域,前者与tRNALeu接受茎结合,后者与MSC的其他相关蛋白相互作用[3-4]。研究表明[21],保守位点T298突变为丙氨酸,则p.T298A影响编辑活性位点的专一性,通过改变活性中心口袋的构象,使正确氨酰化的Leu-tRNALeu被水解而不能累积。D399突变为丙氨酸,则p.D399A的氨酰化活性较野生型的偏低,突变通过远距离影响氨酰化活性中心,导致第一步氨基酸活化反应的效率降低;该突变还改变编辑活性中心口袋的构象,从而影响其水解错误氨酰化tRNALeu的编辑能力。本研究患儿c.1183G > A(p.D395N)位于CP1结构域,蛋白结构预测结果显示,D395突变为天冬酰胺后,不仅改变第395位氨基酸附近氨基酸之间的氢键,同时也影响T298和D399与其它氨基酸之间的氢键,由此可能改变CP1结构域的活性构象,从而影响该区域的编辑功能。此外,c.2133_2135del(p.L712del)突变刚好位于Rossmann折叠结构域的KMSKS肽段(716-720aa)之前,蛋白结构预测结果显示,第712位氨基酸L缺失,破坏709-713和753-755位氨基酸构成的β折叠结构,而KMSKS与第155-164位氨基酸构成的α螺旋结构缠绕在一起,由此影响氨酰化活性中心的功能。两个突变影响了LeuRS蛋白的氨酰化和编辑功能,致使患儿出现LSF1的一系列临床表现。
LSF1临床表现可累及消化道、神经和血液等多个系统[1-2],其发病机制尚未清楚。这种多系统受累的疾病,很容易让人联想到线粒体功能受损。然而Casey等[1]构建了敲除LARS基因的HEK293细胞模型,发现其线粒体的基因、形态及功能均未受影响。有研究[22]发现,LARS基因可以活化mTOR C1(mammalian target of rapamycin complex 1),而mTOR C1可以抑制自噬,当LARS基因缺陷时,mTOR C1的活化受到影响,从而导致机体自噬能力增强。也有研究表明,肝脏mTOR C1的活性减低可导致肝细胞损伤[23]。因此,Casey等[2]提出关于LARS基因突变是通过降低mTOR C1活性使机体自噬能力增强,从而导致多系统受累的设想。
即使LSF1患者仅出现轻度的感染症状,也应当及早干预以防肝衰等严重表现,同时保证有足量的蛋白摄入。然而,Casey等[2]还发现,LSF1患者即使在蛋白摄取充足及肝功能正常的状态下,仍表现有持续的低白蛋白血症。白蛋白中亮氨酸含量最高,虽然LSF1患者的LARS基因突变导致人胞质LeuRS功能受影响,但亮氨酸水平正常,因此补充亮氨酸也并不能改善低白蛋白血症。
本研究分析了第一例非白人ILSF1患儿的临床表现、实验室检查特点。通过外显子组捕获测序和Sanger测序验证,发现患儿为LARS基因新突变c.2133_2135del和c.1183G > A的复合杂合子,确诊为ILSF1,扩展了LARS基因突变谱,同时为患儿诊断及其家庭遗传咨询提供了实验依据。
| [1] | Casey JP, McGettigan P, Lynam-Lennon N, et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy[J]. Mol Genet Metab, 2012, 106 (3): 351–358. DOI:10.1016/j.ymgme.2012.04.017 |
| [2] | Casey JP, Slattery S, Cotter M, et al. Clinical and genetic characterisation of infantile liver failure syndrome type 1, due to recessive mutations in LARS[J]. J Inherit Metab Dis, 2015, 38 (6): 1085–1092. DOI:10.1007/s10545-015-9849-1 |
| [3] | Ling C, Yao YN, Zheng YG, et al. The C-terminal appended domain of humancytosolic leucyl-tRNA synthetase is indispensable in its interactionwith arginyl-tRNA synthetase in the multi-tRNA synthetase complex[J]. J Biol Chem, 2005, 280 (41): 34755–34763. DOI:10.1074/jbc.M413511200 |
| [4] | Cusack S, Yaremchuk A, Tukalo M. The 2 Å crystal structure of leucyl-tRNA synthetase and itscomplex with a leucyl-adenylate analogue[J]. EMBO J, 2000, 19 (10): 2351–2361. DOI:10.1093/emboj/19.10.2351 |
| [5] | Ibba M, Soll D. Aminoacyl-tRNA synthesis[J]. Annu Rev Biochem, 2000, 69 : 617–650. DOI:10.1146/annurev.biochem.69.1.617 |
| [6] | Hu QH, Huang Q, Wang ED. Crucial role of the C-terminal domain of Mycobacterium tuberculosis leucyl-tRNA synthetase in aminoacylation and editing[J]. Nucleic Acids Res, 2013, 41 (3): 1859–1872. DOI:10.1093/nar/gks1307 |
| [7] | Yan W, Ye Q, Tan M, et al. Modulation of aminoacylation and editing properties of leucyl-tRNA synthetase by a conserved structural module[J]. J Biol Chem, 2015, 290 (19): 12256–12267. DOI:10.1074/jbc.M115.639492 |
| [8] | 宋元宗, 牛饲美晴, 盛建胜, 等. Citrin缺陷导致的新生儿肝内胆汁淤积症家系SLC25A13基因突变研究[J]. 中华儿科杂志, 2007, 45 (6): 408–412. |
| [9] | Lu YB, Kobayashi K, Ushikai M, et al. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency[J]. J Hum Genet, 2005, 50 (7): 338–346. DOI:10.1007/s10038-005-0262-8 |
| [10] | Tabata A, Sheng JS, Ushikai M, et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency[J]. J Hum Genet, 2008, 53 (6): 534–545. DOI:10.1007/s10038-008-0282-2 |
| [11] | Kobayashi K, Sinasac DS, Iijima M, et al. The gene mutated in adult-onset type Ⅱ citrullinaemia encodes a putative mitochondrial carrier protein[J]. Nat Genet, 1999, 22 (2): 159–163. DOI:10.1038/9667 |
| [12] | Yasuda T, Yamaguchi N, Kobayashi K, et al. Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type Ⅱ citrullinemia[J]. Hum Genet, 2000, 107 (6): 537–545. DOI:10.1007/s004390000430 |
| [13] | Yamaguchi N, Kobayashi K, Yasuda T, et al. Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations[J]. Hum Mutat, 2002, 19 (2): 122–130. DOI:10.1002/(ISSN)1098-1004 |
| [14] | Zhang ZH, Lin WX, Deng M, et al. Molecular analysis of SLC25A13 gene in human peripheral blood lymphocytes: Marked transcript diversity, and the feasibility of cDNA cloning as a diagnostic tool for citrin deficiency[J]. Gene, 2012, 511 (2): 227–234. DOI:10.1016/j.gene.2012.09.049 |
| [15] | Seiradake E, Mao W, Hernandez V, et al. Crystal structures of the human and fungal cytosolic leucyl-tRNA synthetase editing domains: A structural basis for the rational design of antifungal benzoxaboroles[J]. J Mol Biol, 2009, 390 (2): 196–207. DOI:10.1016/j.jmb.2009.04.073 |
| [16] | Fukunaga R, Yokoyama S. Aminoacylation complex structures of leucyl-tRNA synthetase and tRNALeu reveal two modes of discriminator-base recognition[J]. Nat Struct Mol Biol, 2005, 12 (10): 915–922. DOI:10.1038/nsmb985 |
| [17] | Kobayashi K, Saheki T, Song YZ. Citrin deficiency[M/OL]//Pagon RA, Adam MP, Ardinger HH, et al. GeneReviews. Seattle (WA): University of Washington, Seattle: 1993-2017. 2005 Sep 16 [updated 2014 Jul 31]. |
| [18] | Song YZ, Zhang ZH, Lin WX, et al. SLC25A13 gene analysis in Citrin deficiency: Sixteen novel mutations in east Asian patients, and the mutation distribution in a large pediatric cohort in China[J]. PLoS One, 2013, 8 (9): e74544. DOI:10.1371/journal.pone.0074544 |
| [19] | Burbaum JJ, Schimmel P. Structural relationships and the classification of aminoacyl-tRNA synthetases[J]. J Biol Chem, 1991, 266 (26): 16965–16968. |
| [20] | Chen X, Ma JJ, Tan M, et al. Modular pathways for editing non-cognate amino acids by human cytoplasmic leucyl-tRNA synthetase[J]. Nucleic Acids Res, 2011, 39 (1): 235–247. DOI:10.1093/nar/gkq763 |
| [21] | Pang YL, Martinis SA. A paradigm shift for the amino acid editing mechanism of human cytoplasmic leucyl-tRNA synthetase[J]. Biochemistry, 2009, 48 (38): 8958–8964. DOI:10.1021/bi901111y |
| [22] | Han JM, Jeong SJ, Park MC, et al. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway[J]. Cell, 2012, 149 (2): 410–424. DOI:10.1016/j.cell.2012.02.044 |
| [23] | Umemura A, Park EJ, Taniguchi K, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition[J]. Cell Metab, 2014, 20 (1): 133–144. DOI:10.1016/j.cmet.2014.05.001 |